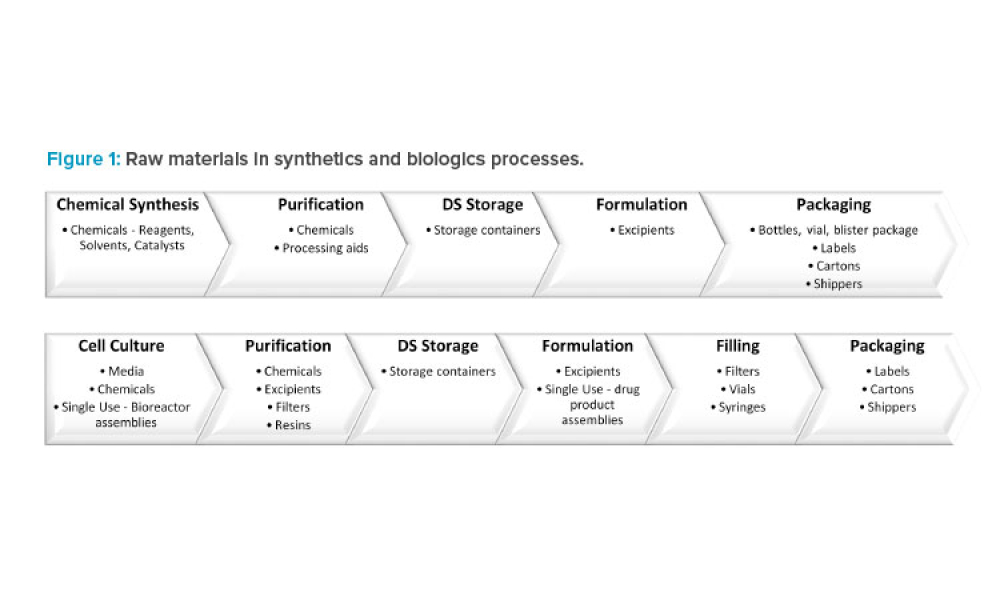
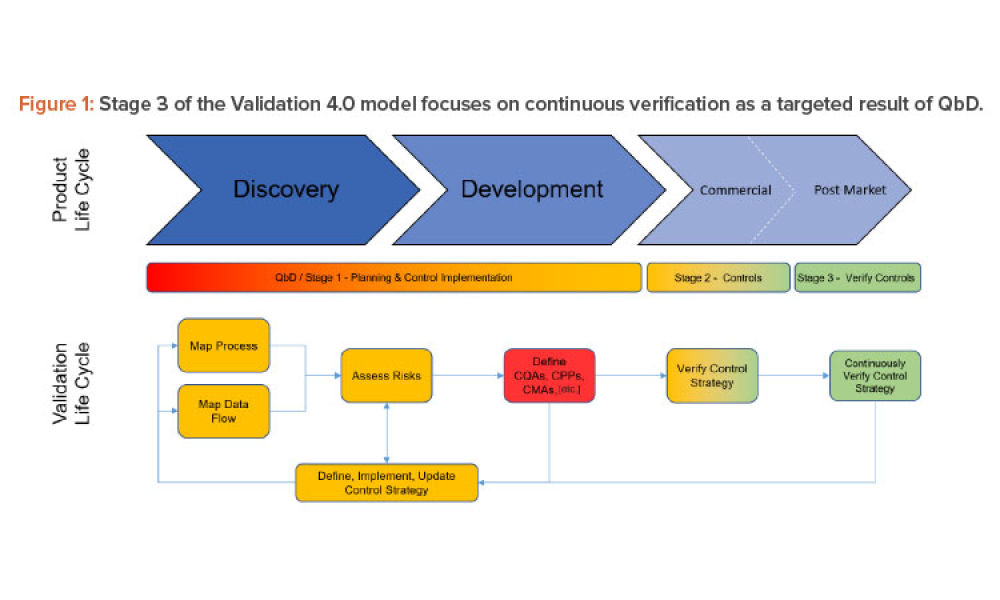
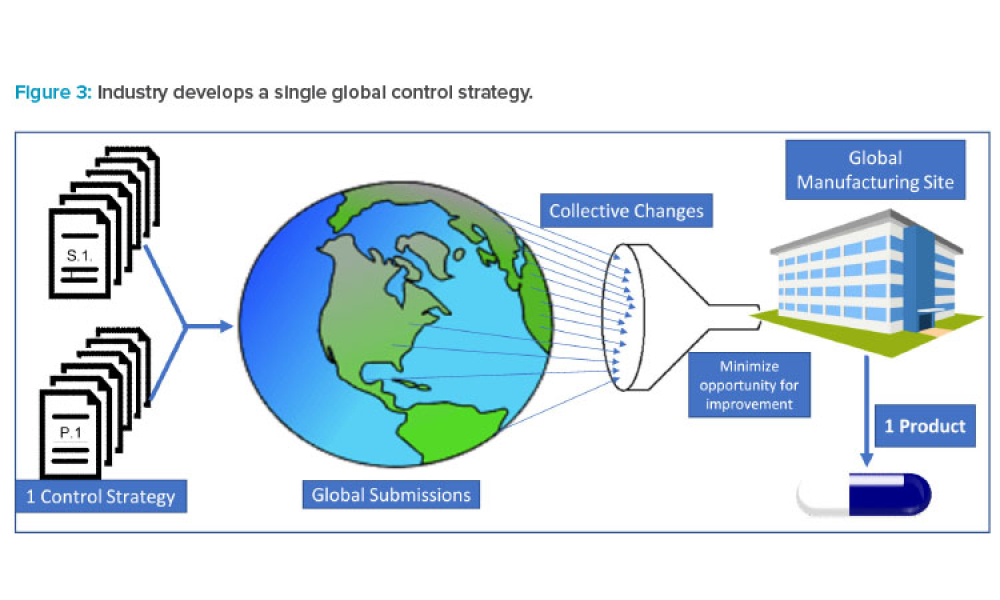
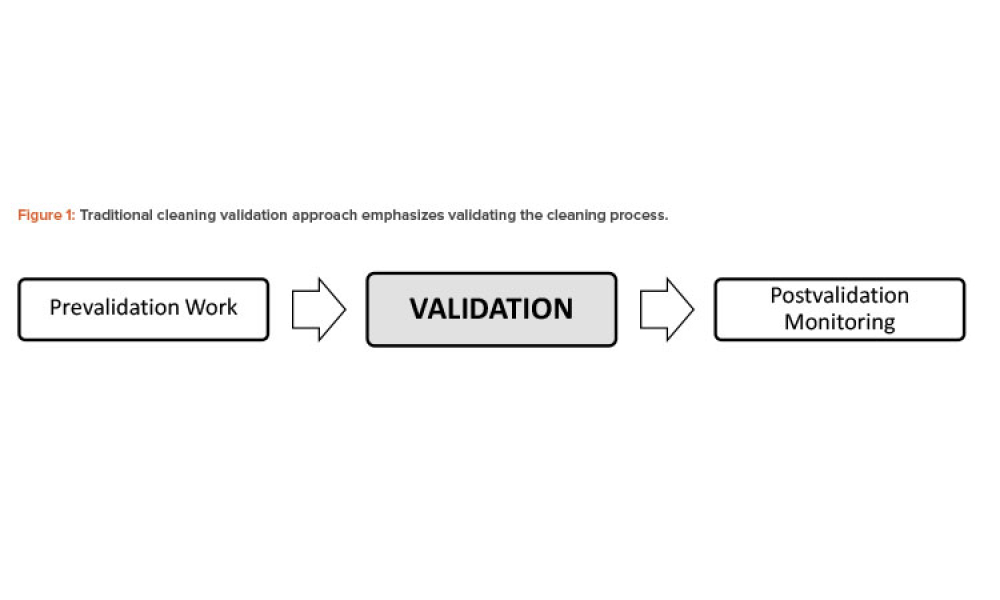
Sponsored Content
Validation professionals are under pressure — from rising workloads to new regulatory demands. Backed by four years of industry data, this year’s State of Validation report — authored by leading life sciences market research Jonathan Kay — reveals how organizations are tackling challenges like audit readiness, resourcing constraints, and the shift to digital systems. Discover benchmarks, best...