Case Study: Facilitating Efficient Life-Cycle Management via ICH Q12

The latest ICH guideline, ICH Q12,
The implementation of Quality by Design as a science-driven, risk-based approach to expand product knowledge and process understanding was intended to serve as a foundation for and encourage continual improvement, and thereby increase assurance of quality for pharmaceutical products. The Quality by Design approach has been a paradigm shift for industry and regulatory authorities because it formally focuses on (a) prospectively characterizing quality risks to patient safety and efficacy, and (b) developing an appropriate control strategy to mitigate those risks.2, 3, 4, 5 Though the adoption of Quality by Design as a development paradigm within the industry has been widely acknowledged as successful, the implementation of Quality by Design to support regulatory applications through the product life cycle was incomplete because there were no provisions for how postapproval changes and improvements would be acceptably submitted and effectively approved. With the advent of ICH Q12, regulatory mechanisms have been introduced to simplify, enable, and expedite postapproval variations and supplements. The appropriate execution of those concepts will validate the principles of Quality by Design. The concept of established conditions has emerged as one of those enabling mechanisms.
The Value of Established Conditions
In conjunction with a robust pharmaceutical quality system (PQS), established conditions describe and present in a regulatory application a comprehensive control strategy for the product through its life cycle. According to ICH Q12, established conditions “are legally binding information considered necessary to assure product quality,” and they reflect a company’s commitment to manufacture and control the drug product and ensure appropriate, consistent, and sustainable quality, safety, and efficacy for the patient.1 In essence, established conditions represent the company’s “license to operate,” by which decisions on postapproval changes are made.
Tools available in ICH Q12, such as the Product Lifecycle Management (PLCM) document, enable the company to distinguish the established conditions from supportive information. The system of risk-based reporting categories also facilitates the use of the Post Approval Change Management Plan (PACMP), which enables predictability in planning for future changes to established conditions. (Note: A Post Approval Change Management Plan is not included in this case study.)
Historically, when planning for a postapproval change to the chemistry, manufacturing, or control (CMC) of a product, the sponsor would have as-sessed all information in module 3 of the common technical document (CTD) with regard to the specific regulatory commitments and postapproval obligations of each relevant region and then would assign region-specific actions. Following the concepts within ICH Q12, all changes—irrespective of the proposed reporting categories—would still be formally assessed using the site change assessment process and managed within the pharmaceutical quality system. The site change management system would continue to ensure that changes are fully documented and progressed through the change management procedure. The change would be assessed to determine its impact on regulatory compliance, quality, and product control strategy, as well as the necessity to revalidate the process. The advantage to implementing the concepts contained within ICH Q12 is that, in accordance with this assessment, the required reporting category for a change to each established condition is defined prospectively in the Product Lifecycle Management.
Given the level of scientific understanding of the process and control strategy, and the rigorous assessment of the impact of postapproval changes on the quality of the drug substance and drug product, some changes to established conditions may be managed within the pharmaceutical quality system without being reported to the regulatory agency. In addition, reduced reporting categories may be scientifically justified, resulting in a shorter time prior to implementation of the change. This prospective life-cycle management planning can enable a company to manage and implement postapproval chemistry, manufacturing, or control changes in a more predictable and efficient manner.
Case Overview
The Prior Approval Supplement (PAS) submission discussed in this case study was included in the US FDA Office of Policy for Pharmaceutical Quality’s established conditions Pilot Program 6 and was submitted less than a year after the initial product approval. The approved original New Drug Application (NDA) for the specific product provided a detailed description of how a science- and risk-based approach was used to define the product control strategy. (The strategy for the product used the enhanced approach per ICH Q8 and Q11,2, 5 along with an explanation of how the regulatory application was aligned with the company's change management system to ensure appropriate quality throughout the product’s life cycle.) The active ingredient was a small molecule manufactured using standard batch process techniques, and the drug product used compendial excipients, direct compression, and a standard tableting process.
During the time when established conditions were prepared for this product, ICH Q12 was at Step 2 draft,7 and the FDA specifically requested Pfizer to include the term “key,” which was subsequently omitted from the final version of ICH Q12.1 Although the term “key” is used in this case study, the choice of established conditions and their respective proposed reporting categories would not have changed if the term had not been used. The outcome, regardless of the term “key,” remains aligned with the intent and guidance provided in the finalized ICH Q12, which recognizes a continuum of risk and criticality.1
In November 2019, the FDA approved the Prior Approval Supplement proposal that defined product-specific established conditions and associated regulatory reporting categories for all aspects of the chemistry, manufacturing, or control commitments in the registration application. The discussion provided in this article is intended to share the strategies used to identify established conditions for this program and the results of a successful interaction with regulators, and to encourage open communication around the opportunities provided in ICH Q12.
The identified established conditions spanned the full scope of manufacturing and included items such as established name, structure, formula, molecular mass, description and composition, batch formula, manufacturing sites, manufacturing procedures, material specifications, critical process steps and intermediates, excipient specifications, release specifications analytical performance, container closure specifications, retest period, and shelf life. The following principles were applied in identifying established conditions and assigning reporting categories, in accordance with the Step 2 ICH Q12 draft:7
- All changes, including those to non-established conditions, were to be managed within the company’s robust pharmaceutical quality system and appropriate documentation was to be available during inspection.
- Established conditions associated with the analytical procedures were defined based on the potential risk to the quality of the product using the knowledge obtained during the development of the methods as well as the sample and its matrix.
- Relationships between process parameters/material attributes and product critical quality attributes (CQAs) were well understood and demonstrated through risk assessments and experimentation.
- All critical process parameters (CPPs) and key process parameters (KPPs) were considered established conditions.
- Changes to established conditions required either prior approval or notification.
Postapproval changes to established conditions required different reporting categories depending on the level of potential risk associated with the change. For each established condition, the risk to product quality associated with a change was quantified through a robust risk assessment, taking into consideration the overall control strategy. The original criticality assessment was completed using two criticality categories: (a) noncritical process parameters or material attributes that do not impact critical quality attributes, and (b) critical process parameters or material attributes that are known to have an impact on a critical quality attribute and require control to ensure quality. Based on a request from the FDA, the criticality assignments were retrofitted to align with the ICH Q12 Step 2 draft guideline.7 In the Prior Approval Supplement, “key” was defined as “a process parameter or material attribute that may have a relationship to a critical quality attribute but has a reduced risk of impacting the safety or efficacy of the product compared with a critical process parameter or material attribute.” This definition was used instead of the definition provided in the Step 2 guideline, which was based on process consistency.
Reporting categories were assigned either in alignment with FDA guidelines8, 9, 10 or justified based on reduced risk to quality supported by appropriate development data. Table 1 shows the linkages for the Prior Approval Supplement of established conditions to criticality/risk to product quality as well as reporting categories for ICH Q12 and several regional guidelines.8, 11 The terminology used for ICH regulatory reporting category was defined in the Prior Approval Supplement in alignment with the ICH Q12 Step 2 guideline:11
- Prior approval (PA): Changes that are considered to have sufficient risk to require regulatory review and approval prior to implementation
- Notification Moderate (NM): Moderate-risk changes that are judged not to require prior approval and generally require less information to support the change
- Notification Low (NL): Minor changes that have minimal potential to have an adverse effect on the identity, strength, quality, purity, or potency of the drug product
- Not Reported (NR): Lowest-risk changes to non-established conditions that do not have a potential impact to quality and are managed and documented only within the pharmaceutical quality system
| EC | Criticality | Approach | ICH Q12 |
United States | European Union |
Japan | Canada | WHO |
|---|---|---|---|---|---|---|---|---|
| Yes | Critical | Implementation after Approval |
PA | PAS | Type II | Partial Change Application |
Supplemental New Drug Submission |
Major Variation |
| Critical/Key | Implementation after Submission and Waiting Period |
NM | CBE-30 | Type 1B | Minor Change Notification |
Notifiable Change | Moderate Variation | |
| Key | Implementation after Submission |
NL | CBE-0 | Type 1AIN | Non-approved Matter |
Level III (annually) | Minor Notification | |
| Submit after Implementation |
Annual Report | Type 1A | ||||||
| No | Noncritical | Managed in the PQS | NR | NR | NR | Level IV | With no impact (on-site/GMP record) |
Typically, changes to noncritical parameters disclosed in the process description require notification (e.g., in the annual report), at a minimum. When following ICH Q12 guidelines,1 all items that are not established conditions do not require postapproval reporting; instead, they are managed within the pharmaceutical quality system. This approach allows operational flexibility and the potential for continual improvement.
Discussion
An abbreviated Product Lifecycle Management document, which was abridged to protect the proprietary nature of the information, is presented in the Appendix to this article (available online, see the box on page 58 with the link). The Appendix captures many of the same elements summarized in the pharmaceutical development control strategy tables. Like the control strategy tables, the Product Lifecycle Management contains the parameters and criteria (many criteria were redacted from the Appendix); beyond that, the Product Lifecycle Management contains associated regulatory reporting categories. Reductions in reporting categories compared to the current US guidelines8, 9, 10 that were justified for this product are highlighted in the following sections.
Drug Substance Manufacturing Process
The drug substance synthesis included three chemical steps from the starting material to the drug substance. The synthesis included an isolation of the Step X intermediate and an isolation and recrystallization of the Step Y intermediate. In addition, there was an isolation and recrystallization of the crude drug substance at Step Z. The three isolations/crystallizations were very efficient at purging a variety of impurities other than the one that was specified in the release specification. To aid the risk assessments, the three chemical steps were further divided into a total of 12 focus areas. Based on the risk assessments, each process parameter was assigned a criticality and associated reporting category. Within the wide ranges in which the process parameters were studied, all parameters that were identified as having a functional relationship with a critical quality attribute were assigned either the “critical” or “key” criticality classification and categorized as established conditions. All process parameters that were identified as having no functional relationship with a critical quality attribute were assigned as noncritical and categorized as non-established conditions.
The Product Lifecycle Management document for drug substance is shown in rows4, 5, 6, 7, 8 of the Appendix. A comparison of the reporting categories in the Product Lifecycle Management versus current FDA guidance8 reveals several instances where the Product Lifecycle Management and the guidelines are aligned, as well as many established conditions that have reduced reporting categories. The latter are described as follows:
- Omission of the recrystallization at Step Y from the manufacturing process will be reported as Notification Moderate (Appendix row 5). The recrystallization of the Step Y intermediate was included in the commercial process as an opportunity to purge impurities. However, data collected to date have demonstrated that the recrystallization is not required to ensure drug substance quality. To improve the efficiency of the drug substance manufacturing process, Pfizer may look to remove the recrystallization via a postapproval submission. The change will be formally assessed using the manufacturing site’s change management process within the pharmaceutical quality system. The process will be revalidated to demonstrate that there is no impact to drug substance quality. Based on this rationale, the removal of recrystallization was accepted as a Notification Moderate, which is a CBE-30 in the United States, as opposed to a Prior Approval Supplement that would otherwise be required based on FDA guidance.8
- Eight critical process parameters will be reported as Notification Moderate rather than prior approval (Appendix row 6). The justification for this downgrade in reporting category is that the control of the process parameters is not the only component of the overall control strategy for the associated critical quality attributes. Other elements of the overall control strategy, such as the drug substance specification, still mitigate the risk. Although these critical process parameters have an impact on a critical quality attribute, the material can be recovered through reprocessing, as allowed in ICH Q7,12 because of the efficient purge of all impurities through the normal crystallization unit operations.
- Three key process parameters that have very low risk to impact quality attributes that are not listed on the drug substance specification will be reported as Notification Low (Appendix row 7). The reporting category was reduced from Notification Moderate because these quality attributes are well controlled by the process and the process parameters’ limits are not the only components of the overall control strategy.
- Twenty-four noncritical process parameters in Steps X and Y will be not reported (NR) but will be managed within the pharmaceutical quality system. Through the enhanced development, it was demonstrated that these parameters lacked both a functional relationship with any critical quality attribute over a wide range and an identified edge of failure. These parameters would otherwise be reported as Notification Moderate based on FDA guidance. 8
- Eighteen Step Z noncritical process parameters that do not impact any critical quality attributes will be not reported but will be managed within the pharmaceutical quality system. These parameters had an absence of a functional relationship with a critical quality attribute over a wide range and an absence of an identified edge of failure. According to the FDA guidance,8 any changes made after the final intermediate processing steps should be reported as prior approval.
Drug Product Manufacturing Process
The film-coated tablets were manufactured using a standard manufacturing process, which included blending, compression, and film coating, and used conventional pharmaceutical manufacturing equipment. The process parameter ranges identified through the development process produced tablets that met the proposed acceptance criteria of the drug product within the operating ranges evaluated. All of the parameters assessed produced tablets that met the acceptance criteria of the specified quality attributes of the drug product.
The manufacturing process remained largely unchanged throughout development, and significant experience at the proposed commercial manufacturing site, on the proposed commercial manufacturing equipment using the same common blend formulation, was available. Blend and lubrication revolutions were designated as critical process parameters based on their potential to affect blend and tablet uniformity. Screen size, film-coat weight gain, and tablet weight and hardness in-process controls (IPCs) were designated as key process parameters based on their low risk or potential relationship to the critical quality attributes. The Product Lifecycle Management document for drug product manufacturing and controls is summarized in the rows 27 and 28 of the Appendix.
A comparison of the reporting categories in the Product Lifecycle Management versus current FDA guidance8 reveals several instances where the Product Lifecycle Management categories are aligned with current guidelines, as well as several examples where reduced reporting categories have been approved:
- For the drug product process description, the reporting categories for screening, blending, compression, and film-coating equipment are aligned with FDA guidance8 if the equipment is changed to a different operating principle (Appendix row 27). Reporting categories for critical process parameters and for the tablet hardness in-process controls are also aligned with FDA guidelines (Appendix row 28).
- Changes to the equipment using the same design and operating principle will not be reported but will be managed by the pharmaceutical quality system change management process. These changes do not have a significant impact on product quality, and this eliminates annual reporting responsibilities for five equipment items (Appendix row 27).
- Changes to tablet weight and film-coating in-process controlss will be reported as Notification Low (Appendix row 28). The downgraded reporting category for tablet weight was justified because weight was monitored throughout compression to allow adjustment if required. The lower reporting category for the film-coating in-process controls was based on extensive prior knowledge with the equipment and coating system at the commercial manufacturing site and because the film coat is nonfunctional. These parameters would otherwise be reported as Notification Moderate based on FDA guidance.8
- Changes to screen aperture will be reported as Notification Low because the screen is used for de-lumping and does not impact particle size. If screening impacted particle size, a change to screen aperture would be reported as prior approval based on FDA guidance.8
Analytical Performance
Regulatory metrics associated with changes to analytical methods in the US are somewhat more nuanced due to the particular verbiage in the FDA guidance on changes to an approved NDA or abbreviated NDA (ANDA),8, which states that alternative analytical methods may be added or revised via an annual report (a Minor Change) as long as this “alternative analytical procedure […] provides the same or increased assurance of the identity, strength, quality, purity, or potency of the material being tested as the analytical procedure described in the approved application.” This represents a useful path for industry as a way of making modifications to methodology via a low reporting category. However, for purposes of determining compliance, regulatory analytical procedures (i.e., not alternative procedures) are used. Consequently, the unaltered analytical procedure is still “on the books.” Furthermore, designation of an alternative analytical procedure as a regulatory procedure is categorized as a Major Change, requiring a Prior Approval Supplement. The FDA guidance8 indicates that changes to an approved regulatory procedure can be prosecuted, by inference, via a CBE-30, as long as the revised procedure provides the same or increased assurance of the identity, strength, quality, purity, or potency of the material being tested.
The established conditions for the purity methodology to support the quality evaluation are outlined in rows 17–21 of the Appendix. The established conditions ensure the methodology will effectively separate and quantify specified degradation products in the drug substance. The purity method is a standard reversed-phase high-performance liquid chromatography (HPLC) procedure using a C18 column and an acetonitrile-buffer solvent system with gradient elution and ultra-violet (UV) detection. The analytical methods to assess the purity of the drug substance and drug product included established conditions based on the method principle, method-specific performance criteria, and higher-level method parameters. This performance-based approach was grounded on enhanced understanding of the method and the sample matrix. The established conditions were focused primarily on performance attributes of the method (e.g., validation criteria per ICH Q2), but they also included key method parameter acceptable ranges rather than set points. This set of established conditions will ensure that any method revisions will result in performance that continues to be aligned with the requirements of the method. Although not all method parameters are reported as established conditions, parameters are selected to provide a boundary for what parameter changes may be implemented within the company’s pharmaceutical quality system. Defining the performance criteria as established conditions further ensures any minor parameter changes will provide equivalent or better results than the original methodology.
By indicating specific categories of changes as defined in the Product Lifecycle Management document, the use of established conditions helps alleviate whatever ambiguity these multiple options for prosecuting changes impart in terms of the appropriate reporting category to be used. In addition, by applying science- and risk-based assessments of the nature of the change, it is possible to downgrade the reporting category significantly, in some cases to the point where regulatory notification is not required. This can be illustrated through the following analysis. For the purposes of this analysis, it is assumed that the desire is to revise the approved regulatory analytical procedure, not merely introduce an alternative procedure. Six analytical procedures were included in the Prior Approval Supplement that de-fined established conditions. They included three liquid chromatography procedures, one using gas chromatography, one using laser diffraction for particle size, and one for dissolution of the drug product with UV end analysis.
- For each of the six procedures, the method principle (e.g., reversed-phase chromatography or spectroscopy) and the validation criteria as outlined in ICH Q213 were defined as established conditions, with the highest risk associated with the quality of the product. The method principle and performance criteria, a total of 65 established conditions in all, will be reported as prior approval.
- For each of the six procedures, several of the higher-level operational parameters (e.g., solvent system for HPLC) and the system suitability criteria, 35 established conditions in all, will be reported as Notification Moderate (i.e., CBE-30); this is consistent with the FDA guidance,8 in that changes could be made to these parameters and criteria while still maintaining the same or increased assurance of the identity, strength, quality, purity, or potency of the material being tested as the approved analytical procedures.
- Six slightly more detailed parameters (e.g., the wavelength of the analysis) were still considered established conditions but will be reported as Notification Low (i.e., in an annual report). Because these parameters would otherwise have been reported as Notification Moderate based on FDA guidance,8 they represent additional flexibility.
- As outlined previously, changes to non-established conditions (65 parameters) are handled entirely within the pharmaceutical quality system, with no reporting to FDA necessary. These parameters would otherwise have been reported as Notification Moderate based on the FDA guidance; therefore, they represent the largest component of additional flexibility in the area of the analytical procedures.
Specifications
A safety-based approach was used to identify reporting categories for specification established conditions to support the overall control strategy. The overall control strategy for ensuring product quality relies on upstream specifications established in conjunction with process understanding. For example, an impurity may have a specified limit in a drug substance starting material at a level that is known to be well purged by the first two steps of the drug substance manufacturing process. Given the purge knowledge, this impurity and its fate product may not be listed on downstream specifications (e.g., intermediate or drug substance) in addition to the starting material specification. The established condition reporting categories for upstream specification limits (starting material and intermediate) are therefore based on how a change to that established condition would ultimately impact the quality of the drug substance and/or drug product.
The reporting categories for changes to the quality attributes listed on the drug substance specification largely align with the FDA guidance on changes to an approved NDA or ANDA.8 However, a safety- and risk-based approach was approved for changes to impurity established conditions, as shown in the Appendix row 16. For this case study, the drug is approved for treatment of a specific subset of patients with metastatic non-small-cell lung cancer (NSCLC), an indication that falls under the scope of ICH S9.14 Both ICH Q3A15 and ICH S914 guidance on development of anticancer pharmaceuticals provide for some modification of control strategies to be proposed for such pharmaceutical products.
- Because the drug substance is used for the treatment of advanced cancer, quantitative structure-activity relationship (QSAR) findings for potential genotoxicity would not require low-level controls. Given this safety-based risk assessment, impurities in the range of 0.10% to 0.15% can be added to the specification via Notification Moderate with appropriate validation data in line with the established conditions. If an impurity above 0.15% were to be added to the specification, the appropriate supporting toxicology package would be assembled and submitted with the proposed specification change via a prior approval mechanism.
- Using a safety- and risk-based reporting approach, an increase to an impurity limit in a starting material or the addition of a new impurity to the starting material specification would not necessarily require prior approval to implement, as shown in row 10 of the Appendix. If it is demonstrated that raising an impurity limit in a starting material does not result in a change to drug substance quality, that starting material limit change should be made via a Notification Moderate, with the relevant supporting data. Changes to starting material limits that would also result in a necessary change to the drug substance specification would be filed as PA in conjunction with the drug substance specification change.
- A similar approach was taken with the raw material specifications, as shown in row 11 of the Appendix. Special consideration was made for established conditions that could impact the safety of the drug substance (e.g., benzene levels from solvents or palladium levels). If a change to a raw material specification would not impact the quality or safety of the drug substance, the reporting category is lowered to Notification Moderate.
- A similar safety- and risk-based approach was used to define reporting categories for critical in-process controls and intermediate specifications, as shown in row 12 of the Appendix. The reporting category ultimately depends on the impact that change has on the quality of the drug substance. Additionally, if it is understood that the process control limit is not critical for ensuring drug substance quality (it may be referenced as “key”), the reporting mechanism was lowered to Notification Low.
- The quality attributes listed on the drug product specification are identified as critical established conditions. There were no specific change categories defined for these attributes because they are aligned with the recommendations in the FDA guidance,8 as shown in row 31 of the Appendix.

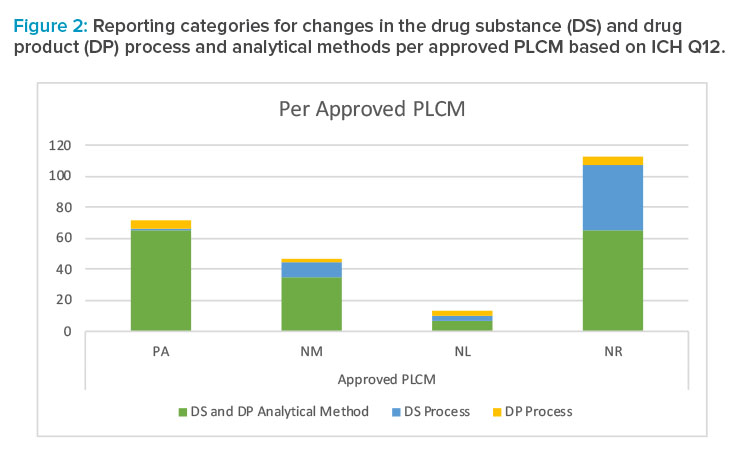
Summary of Reporting Categories
The difference in reporting categories according to the FDA guidance8 versus the approved Product Lifecycle Management is apparent when comparing the graphs in Figures 1 and 2. The largest impact of applying the concepts in ICH Q12 was the ability to manage the 65 individual detailed analytical method operational parameters, 42 drug substance process parameters, and five drug product process parameters within the pharmaceutical quality system without reporting. It is clear that the highest degree of regulatory flexibility was achieved for potential changes related to (a) the analytical method parameters, where the number of changes requiring regulatory review and approval (Prior approval and Notification Moderate) was reduced from 172 to 100, and (b) the drug substance manufacturing process parameters, where the number of changes requiring regulatory review and approval (Prior approval and Notification Moderate) was reduced from 32 to 11. This operational flexibility should encourage both innovation and continual improvement and improve proactive planning of supply chain adjustments.
The largest impact of applying the concepts in ICH Q12 was the ability to manage the 65 individual detailed analytical method operational parameters, 42 drug substance process parameters, and five drug product process parameters within the pharmaceutical quality system without reporting.
Conclusion
ICH Q12 [1] provides the regulatory framework to facilitate continual improvement and bridge the technical and regulatory gaps that prevented the postapproval flexibility sought by applying Quality by Design concepts. Established conditions introduce provisions for reducing the life-cycle management burden and decreasing the time needed to implement some postapproval changes, while at the same time providing quality assurance throughout the product life cycle. The significant reduction in required regulatory reporting for postapproval changes that have low risk to product quality will allow more effective use of resources for both industry and regulatory agencies. In this case study, the approved Product Lifecycle Management does not require regulatory submission for 112 parameters for which postapproval changes would otherwise have been reported, and these parameters represent the largest increase in flexibility gained through the approval of the established conditions listed in the Product Lifecycle Management. Strategies with potentially high impacts include (a) the reduction in reporting category from prior approval to Notification Moderate for the omission of one of the recrystallization steps in the drug substance process; (b) the ability to manage the 65 individual detailed analytical method operational parameters in the pharmaceutical quality system; and (c) the agreement on the safety- and risk-based approach for changes to impurity established conditions, which allows impurities in the range of 0.10% to 0.15% to be added to the drug substance specification via Notification Moderate rather than prior approval. It should be noted that products developed using minimal or traditional approaches (i.e., not enhanced) may not result in the same level of flexibility through the ICH Q12 1 framework as those developed through an enhanced approach.
Regulatory approval of established conditions by the US FDA permits some future postapproval changes for this product to be managed within the company’s pharmaceutical quality system with-out regulatory reporting. Global change implementation planning will be complex as regulatory authorities in other jurisdictions may still require postapproval regulatory submissions. In addition, alignment of local regulatory reporting categories with ICH Q121 is needed; this is especially an issue in regions where changes in the legal framework are required. Global pursuit and acceptance of this endeavor is critical to realize the value of the ICH Q12 vision to harmonize life-cycle management and reduce the postapproval regulatory burden. Postapproval reporting requirements that focus on product quality, safety, and efficacy significantly reduce the number of postapproval submissions and encourage and facilitate continual improvement.
Appendix: Abridged Product Lifecycle Management from the Full Product Lifecycle Management Approved in the PAS
An appendix presenting an abridged version of the Product Lifecycle Management approved in the PAS is included in this online version.
About the Authors






Acknowledgements
The authors are grateful to the Pfizer colleagues from across Pharmaceutical Sciences, Global chemistry, manufacturing, or control, and Pfizer Global Supply for their input in setting the established conditions and various helpful discussions in the development of the strategies discussed in this article. A special thank you to Neil Clayton, Graham Cook, Olivi-er Dirat, Timothy Graul, John Groskoph, Sylvia Hoff, Margaret Howard, Karen Kelly, Sree Malepati, Marie O’Brien, Ron Ogilvie, Gerald Segelbacher, and Cindy Wechsler.


