Regulatory Landscape for Raw Materials: CMC Considerations
A reliable supply of raw materials is critical to maintain a robust supply chain to serve patients globally. With shortages, regulatory complexity is compounded due to differences in submission and data requirements from various regulatory agencies. Therefore, there is an increasing need to implement a harmonized regulatory infrastructure that is both flexible and predictable to provide more agility without product delays.
The pharmaceutical manufacturing supply chain starts with the raw materials, which are needed to ensure drug availability for patients. With ever-increasing supply chain challenges, raw material shortages have become a point of discussion. In this article, the term “raw material” refers to a material used in the manufacturing and packaging of a drug substance (DS) or a drug product (DP).
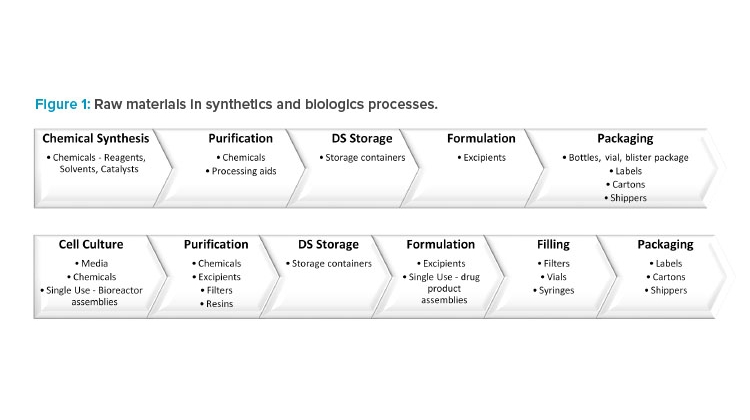
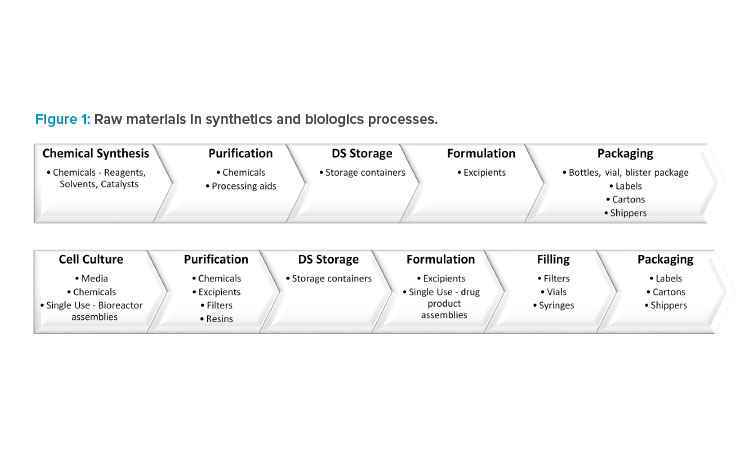
For a synthetic drug, the DS is chemically synthesized in multiple ordered steps from the starting materials using a range of chemicals. This is followed by DP manufacturing, where the DS is formulated with excipients. Finally, the DP is packaged in a suitable container to ensure continued quality.
For a biologic drug, the DS is manufactured upstream in cell culture media followed by downstream purification, which requires chemicals, filters, and resins. The DP formulation and filling processes use excipients, filters, vials, and syringes. In addition, single-use technologies have been increasingly employed throughout manufacturing because of the advantages they offer, including reductions in cost, manufacturing footprint, contamination risk, and processing times (Figure 1).
Although they have been historically overlooked as a key element, raw materials are a critical component at every stage of the drug manufacturing processes. Recent US FDA data show that the lack of raw material availability contributes to 27% of drug shortages (see Appendix).
There is undoubtedly a need for improved supply chain flexibility to address shortages. In cases where raw materials are single sourced, supplier manufacturing problems or product facility closures could result in manufacturing delays and/or stoppages. Similarly, an increased demand forecast could lead to a raw material shortage. One possible mitigation strategy is to build sufficient inventory to ensure continuous product supply. However, large inventories increase the cost of production and the risk of scrapping raw material lots that exceed their shelf life before they can be used.
Diversification and redundancy of raw material supplies by qualification of new raw material sources ensure a geographic footprint of manufacturers providing flexibility and supply resiliency. However, use of alternative raw materials may require approvals from multiple health authorities. Waiting for approvals can significantly delay implementing a change, and the timelines vary between regions, adding further complexity to supply management. For example, implementation of an alternative vial would typically require 4 to 6 months for approval in the EU and US but more than 18 months in other countries. In some cases, to meet the forecast, DP manufacturers manufacture at risk while waiting for approvals for second-source supply.
During the pandemic, the pharmaceutical industry faced challenges in the production of COVID-19 therapeutics and vaccines to meet global demand, as well as mitigation of drug shortages for non-COVID-19-related products, without compromising product quality or patient safety. Lessons learned during the pandemic could be leveraged for future procedures and regulatory submission requirements. This article highlights the regulatory expectations of raw materials, the challenges of postapproval changes. and the impact on supply resiliency. Case studies are presented that demonstrate the importance of defining the raw material attributes that are critical to product quality and how this could support increased postapproval flexibility (including the use of ICH Q12 principles).
Regulatory Expectations
The International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) guidelines contain information regarding regulatory requirements for raw materials. There should be a system for evaluating critical suppliers and a specification agreed upon with the supplier and approved by quality. Upon receipt, incoming raw materials should be tested against specifications that include critical attributes, analytical procedures, and acceptance criteria. Additional requirements are described in ICH Q7.1 The Common Technical Document (CTD) for the Registration of Pharmaceuticals For Human Use: Quality—M4Q guidance covers the minimum requirements for submission of raw materials; however, certain regions have additional requirements. 2
Raw materials used in the manufacture of the DS should be listed in CTD section 3.2.S.2.3, Control of Materials. The name of each material, where it is used in the process, and information on the quality and control should be provided. The material manufacturer is not required for all cases but is often requested by some health authorities for critical materials such as filters. A compendial or multicompendial grade should be listed where applicable; for all noncompendial materials, specifications should be included. Information demonstrating that the quality of the raw materials meets standards appropriate for their intended use should be provided. For example, biologically sourced raw materials may require careful evaluation to establish the presence or absence of deleterious endogenous or adventitious agents.
Per ICH Q11, the potential for material attributes that impact DS critical quality attributes should be identified.3 Raw materials used near the end of the manufacturing process have greater potential to introduce impurities into the DS than raw materials used upstream; therefore, tighter control of quality should be evaluated. A risk assessment to define the control strategy of raw materials can include an assessment of manufacturing process capability, attribute detectability, and severity of impact. For example, the ability of the DS manufacturing process to remove an impurity or limitations in detectability (e.g., viral safety) should be considered. The risk related to impurities is typically controlled either by raw material specifications or robust purification steps later in the synthesis.
An excipient is formulated with the active pharmaceutical ingredient and is typically not chemically or physically altered prior to use; therefore, all components are likely present in the DP. The intended end use of the excipient should be considered when determining the appropriate regulatory and GMP requirements for the excipient and its manufacturing facility. The quality of the excipients and the container/closure systems should meet pharmacopeial standards, where available and appropriate. Otherwise, suitable acceptance criteria should be established. The use of a noncompendial mate-rial may be considered acceptable with strong scientific justification. For a multicompendial excipient that may be marketed for global use, the DP manufacturer should demonstrate conformance of the excipient to the monograph requirements found in specified compendia.
A description of the DP and its composition is provided in CTD section 3.2.P.1, Description and Composition of the Drug Product. More details regarding the quality of excipients are provided in CTD section 3.2.P.4, Control of Excipients. For the European Medicines Agency (EMA), functional related attributes should also be considered, and it may be necessary to include additional tests and acceptance criteria, depending on the intended use of the excipient (see Appendix). For excipients of human or animal origin, information should be provided regarding adventitious agents in CTD section 3.2.A.2, Adventitious Agents Safety Evaluation. For novel excipients (i.e., excipients used for the first time in a DP or by a new route of administration), full details of manufacture, characterization, and controls, with cross-references to supporting safety data, should be provided according to the DS format in CTD section 3.2.A.3, Novel Excipients.4
Additionally, excipients and primary container components may be subject to regional regulatory requirements. For example, the National Medical Products Administration (NMPA) requires registration of high-risk excipients and primary container components using a master file that is referenced by the DP sponsor.
Postapproval Change Management
When a drug manufacturer intends to introduce a change, the potential impact on the process and product quality must be assessed. 1, 5, 6 A change is classified as major, moderate, or minor depending on its nature and impact. A major change is one that requires submission and approval by a health authority prior to distribution of post-change material. A moderate change is one that typically requires submission to a health authority but may not require approval prior to distribution of post-change material. A minor change is reported to the health authority after implementation and does not require a submission prior to product distribution. The classification helps determine the data required to demonstrate comparability (pre- and post-change) and confirm no adverse impact on product quality.
A formal change control system under the company’s pharmaceutical quality system (PQS) is required to evaluate all raw material changes, with established procedures for identification, documentation, review, and approval. A quality risk management system provides assurance to the health authorities that the applicant can ensure process consistency and product quality while continuously monitoring, verifying, and mitigating identified risks. After approval and implementation of the change, there should be an evaluation of the first batches produced post-change.
Health authorities have divergent classifications for changes in terms of risk to product quality and documentation/data requirements. Table 1 shows the classifications assigned (based on published guidance) to three distinct types of raw material changes for biologics (B) and synthetics (S) across six regulators (FDA, EMA, Health Canada, Therapeutic Goods Administration [TGA], Pharmaceuticals and Medical Devices Agency [PMDA], and NMPA) and the World Health Organization (WHO):
- Relaxing acceptance criteria or deleting a test for a raw material. Although this change is not explicitly described in the TGA guidance, a change category requires that any change to raw material specifications be submitted as a Category 3 application requiring prior approval. The PMDA classifies such a change as a partial change application requiring prior approval if the acceptance criteria or test is registered in M1.2. It is considered a moderate change by the FDA (CBE30) and NMPA. In the EMA, Health Canada, and WHO, such a change would be considered minor, provided the deleted parameter was redundant or obsolete. In the case of deletion of an attribute specification that may have a significant effect on product quality, the EMA classifies it as a major type II variation requiring approval before implementation. Health Canada classifies this type of change as level 2 for biologics, which requires approval prior to implementation, or level 3, which requires immediate notification for synthetics, which allows implementation prior to reporting to the agency.
- Relaxing acceptance criteria for compendial excipients to comply with changes to compendia. This change ranges from a moderate change (CBE-30) by the FDA to a minor change not requiring prior approval by the WHO.
- Change to manufacturer or supplier of excipients or raw materials. Classifications vary widely by region depending on the raw material involved and route of administration. Consistently a change in the source of an excipient to one that carries a risk for transmissible spongiform encephalopathy (TSE) is considered a major change. This classification can be reduced to a minor change according to Health Canada and the WHO if supported by a valid TSE Certificate of Suitability (CEP).
Some health authorities do not include all three changes described in their postapproval guidance; for example, the FDA provides guidance for synthetics, but not biologics. Changes not covered need case-by-case management. In addition, submission categories vary between health authorities, making it very challenging to manage the submissions for a raw material change globally. In some guidance documents, changes require associated conditions to be met and documentation/data to be provided in a specified submission category. If a condition cannot be met, then the submission category may be upgraded to a higher category.
Additional examples of postapproval changes for FDA, EMA, Health Canada, TGA, PMDA, NMPA, and WHO are described in detail in the Appendix. The categories in the Appendix assume all conditions are met, required documentation is available for submission, and they are aligned with health agency expectations. The absence of any of the listed documentation should be scientifically justified.
Due to global regulatory requirements, many postapproval changes cannot be implemented until the health authorities have reviewed and approved the change, which can take considerable time. During technical review, additional time and resources may be required to address requests for information from agencies. Because of the lack of harmonization across regions, it is difficult to predict the time that it will take for approval by each health authority. The estimated global approval times for major changes vary considerably—from less than 6 months in some major markets to greater than 18 months in others—resulting in periods of several years before full global implementation of a change can occur.7
This results in a lack of supply chain agility to implement changes when faced with immediate supply shortages. Managing a strategy to accommodate varying global approval timelines is a challenge. Similarly, there are regulatory hurdles to implementing raw material improvements postapproval to proactively improve raw material reliability (e.g., innovative technologies and raw material specification changes enabled through scientific understanding of raw material attributes and their impact on product quality).
Addressing Challenges for Postapproval Changes
Multiple asynchronous reviews of the same information with varying approval timelines across global health authorities result in a more complex supply chain, without improving safety, quality, or efficacy. Currently, a streamlined data package for fast global implementation of a change is unlikely to be accepted due to differing regional data requirements.
The implementation of a global regulatory infrastructure that is harmonized, flexible, and predictable would provide drug manufacturers the agility to expedite raw material supplier qualifications to be better equipped to face raw material challenges while maintaining product quality and supply to patients. The identification of the critical raw material attributes and appropriate setting of specifications is a crucial first step.
| Health authorities |
Relaxing acceptance criteria or deleting a test for a raw material |
Relaxing acceptance criteria for compendial excipients to comply with changes to compendia |
Change to manufacturer or supplier of excipients or raw materials |
|---|---|---|---|
| FDA | Moderate CBE-30
|
Moderate: CBE-30
|
Annual Report
|
| EMA | Major Type II
Minor Type IA
|
Major Type II
Minor Type 1B
|
|
| TGA | Category 3
|
Notification
|
Category 3
Self-Reportable
Notifications
|
| Health Canada | Level 2 Notifiable Change
|
Level 3 Annual Notification (Minor)
compendial raw material to comply with an updated pharmacopeial standard/monograph is Level 4: Not reported (B) |
Level 1 Supplement (Major)
|
| PMDA |
Partial Change Application
|
No Impact
|
No Impact
|
| NMPA |
Moderate
Note: Changing the specification of an excipient where the quality control level is not lowered is also moderate (S) except when tightening quality control limits is a minor change (S). In comparison, addition of test item or tightening of limit of specification is moderate for biologics (B) |
Not described in the guidance | Major
|
| WHO |
Minor Quality Change
|
No Impact
|
Major Quality Change
|
Attribute-focused Approach to Developing Material Specifications
A robust raw material control strategy can be achieved with an attribute-focused approach to identify critical material attributes. This approach facilitates the development of science-based raw material specifications and phase-appropriate decisions across the life cycle of a material. It is important to engage in material attribute understanding early in commercial process development when raw materials are being selected. A well-defined material target profile can be used to conduct a material attribute assessment, and based on that profile, a control assessment can be completed. This can be executed in several stages:
- Define the role of the raw material. Determine how it will be used in the process and what functions it needs to perform its intended use.
- Assess the attributes that the raw material requires to perform the desired function and identify the critical material attributes that impact the process performance and product quality.
- Define the desired target and allowable range for each material attribute based on the knowledge and understanding of the process tolerance.
- Build a control strategy to define the material attribute controls required, from the raw material manufacturing to the receipt and testing at the drug manufacturer.
The attribute-focused approach enables identifying critical material attributes and developing science-based specifications, which are established based on the intended use of the material and the process requirements; for example, avoiding the use of compendial-grade specifications when noncompendial material will suffice or avoiding the use of technical-grade raw materials when more control is required. In addition, having clear user requirements facilitates more informed supplier selection and can support the identification of established conditions (ECs) for raw materials in regulatory filings.
Once the critical material attributes have been established, specifications defined, and suppliers onboarded through the pharmaceutical manufacturer's quality management system, raw material performance can be monitored using attribute data analytics. This enables the predictive assessment of raw material variation, identification of the source of variability, and implementation of proactive mitigations strategies to prevent failures.7
Regulatory submissions preferably include only the critical material attributes. For postapproval raw material changes, the material target attribute profile can facilitate a strong scientific justification based on the knowledge and understanding of the process and the critical material attributes. Some examples of noncritical details include registering trade names, listing part/catalog numbers, and information included in the supplier certificate of analysis that is not relevant to ensure product quality. Registration of these details may limit options of second sourcing, especially in the worst-case scenario when a supplier discontinues a material.
Utilization of Regulatory Tools in ICH Q12
ICH Q12 helps streamline postapproval change implementation by establishing harmonized change categorization, including the identification of the portions of an application requiring a submission if changed postapproval.8 The level of submission category for a change is determined by the level of risk associated with making the change. ICH Q12 provides a framework to enable the modification of some submission categories for changes based on scientific understanding and the level of risk associated with the change.
It includes regulatory tools such as ECs, postapproval change management protocols, and the product life-cycle management document to enhance the manufacturer’s ability to manage chemistry, manufacturing, and controls (CMC) changes effectively under the company’s PQS.9 Adoption of the principles of ICH Q12 could result in fewer postapproval submissions and the ability to implement more changes prior to notification.
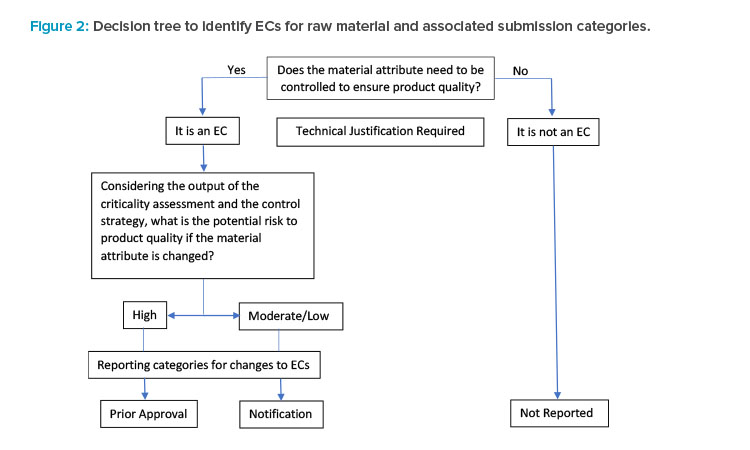
According to ICH Q12, “ECs are legally binding information” within an application considered necessary to assure product quality. Any change to an EC requires a submission to the health authority. Identifying ECs enables a risk-based framework, allowing the use of scientific knowledge and risk mitigation to justify the submission category of a change.
The number of ECs for a raw material, how narrowly they are defined, and the associated submission category depend on several factors:
- Characterization of the product and detection limits of product quality attributes: Development approach adopted, which dictates the level of process and product quality understanding.
- Performance based: High level of scientific understanding of the material attributes that have an impact on process performance and product quality. Data-driven enhanced control strategy primarily focused on the control of process outputs and an improved understanding of the risk.
- Parameter based: Limited understanding of relationship between inputs and resulting product quality attributes. A larger number of material attributes are considered potentially critical.
- The potential risk to product quality when implementing changes to the EC: Risk assessment activities should follow approaches described in ICH Q9 and must consider the overall control strategy and any possible concurrent changes. 10
In general, enhanced knowledge and understanding of the relationship between raw material attributes, process parameters, and product quality enable the identification of parameters critical to product quality, leading to a reduction in the number of ECs. For example, employing a performance-based approach to development can demonstrate that a material attribute that was initially considered potentially critical (in a parameter-based approach) is not actually critical and has no impact on product quality.
A decision tree (Figure 2) was modified from ICH Q12 that illustrates the stepwise approach to identifying ECs for raw material attributes and the as-sociated submission categories (in the context of process parameters). For parameters that are not ECs, postapproval changes are not reported.
Overall, agreement with regulators on the ECs and associated submission categories can reduce the number of postapproval submissions to only the changes most critical to ensuring product quality. This provides more flexibility to implement changes and thus the ability to react more quickly to supply chain challenges. In the long term, a collaboration between regulators and industry stakeholders to develop and implement harmonized guidelines for raw materials would help address flexibility challenges, prevent delays in implementing process improvements, and ensure that both regulator and industry resources are devoted to the most critical issues.
Case Studies
This section describes case studies of postapproval changes to raw materials and the regulatory challenges. The examples highlight the value of well-characterized raw materials and the importance of only including critical material attributes in regulatory submissions. They are representative of issues manufacturers face when attempting to address supplier and quality aspects of raw materials.
Case Study 1: Polypropylene Glycol—Removal of Noncritical Attribute from Specification
The original molecular weight (MW) specification for polypropylene glycol 2000 (PPG) of 1800–2200 was based on the Food Chemical Codex monograph (90%–110% of label) and not based on a scientific understanding of the process/product requirements. By employing an attribute-focused approach, an assessment of MW was performed based on a review of literature, process understanding, process performance, and historical PPG release testing data. The analysis showed no correlation between antifoam performance and MW, and a wider MW range of 1200–3000 was deemed acceptable for use in the processes. Based on the process performance and robustness of the raw material supply quality, it was concluded that the MW attribute is not critical and can be removed from the PPG specification to reduce the business risk without impacting the quality of the DS.
| Description | Polypropylene glycol, average molecular weight of 2000 Daltons | |
| Intended function | Defoamer | |
| Required characteristics to perform the intended function |
|
|
| Material attribute | Target ranges | Justification/control strategy |
| Appearance | Colorless to almost colorless liquid |
Basic GMP requirement tested for each batch–confirms correct material received and may be indicative of impurities present. |
| Identification | Pass/conforms | Basic GMP requirement. Raman, infrared, or near-infrared tested for each batch–confirms correct material received and may be indicative of impurities present. |
| Average molecular weight |
1200–3000 | No correlation between PPG MW and process performance or product quality. Historical quality control (QC) data was 1875–2509 and does not trend close to upper or lower range of 1200–3000, demonstrating robustness of supply and that supplier controls ensure MW inside acceptable range. |
| Density | 0.985–1.014 | No impact |
| Refractive index | 1.450–1.452 | No impact |
| Water | ≤ 0.1% | No impact |
| Viscosity | 400–500 MPAS (20°C, neat) |
No impact |
| Acid value | 0.00–0.08 mg KOH/g | No impact |
| Hydroxyl value | 40–60 mg KOH/g | Supplier release specification includes hydroxyl value which correlates to the average MW (average MW 1200–3000 corresponds to hydroxyl value 37.4–93.5). Historical hydroxyl value from manufacturer have ranged from 53.5 to 56.4 (the supplier acceptable range 40–60). |
| Description | Betaine | |
| Intended function | Protects cells from high medium osmolarities by providing binding sites for both positively and negatively charged species (thereby reducing osmolarity). Reduces the fraction of high-mannose oligosaccharide species. |
|
| Required characteristics to perform the intended function |
Be present in sufficient quantity to achieve target concentration in process. | |
| Material attribute | Target ranges | Justification/control strategy |
| Appearance | White to off-white powder | Basic GMP requirement tested for each batch–confirms correct material received and may be indicative of impurities present. |
| Identification | Pass/conforms | Basic GMP requirement. Raman or infrared tested for each batch–confirms correct material received and may be indicative of impurities present. |
| Water | ≤ 3% | No impact to process or product:
for hygroscopic materials. |
| Assay | ≤ 98% (anhydrous basis, titration with HClO4) | No impact |
At the time of assessment, removal of the MW specification could be reported without requiring approval in the US and Canada and required prior approval in four regions: Australia (3 months for approval), EU (up to 6 months for approval), China (up to 10 months for approval), and Israel (required EU approval first, up to 1 year for approval). The same rationale for the change was submitted globally.
Case Study 2: Betaine—Widening of Raw Material Specification Criterion
Betaine has no compendial monograph, and the original specification included water with an acceptance criterion of ≤ 2.0%. It is a hygroscopic material that transitions to the monohydrate form on absorption of water. This results in water uptake during standard material handling and a risk of failing in-coming quality control testing for the water content attribute.
A technical assessment was performed, demonstrating that increased water content is not expected to have any impact on process or product quality. Based on the chemical properties of betaine and its functional use in the process, a specification of ≤ 3% for water content was considered appropriate. In addition to specification changes, several mitigations were put in place regarding material handling.
At the time of assessment, widening of the water specification was reportable as a notification in Australia, China, and Canada. For many markets, this change did not require reporting to the health authority. This is an example of a change involving a well-characterized raw material resulting in shorter timelines to implementation.
Case Study 3: Sodium Deoxycholate—Removal of Noncritical Attribute from Specification
Sodium deoxycholate is a noncompendial white crystalline powder manufactured by neutralizing deoxycholic acid with sodium hydroxide (NaOH). The amount of NaOH added during the raw material manufacturing determines the conversion to the more soluble sodium salt and the pH in solution. The pH specification for a 10% solution was set at 8.2–10.0 to avoid precipitation at values below 8.2 caused by residual deoxycholic acid.
It was recognized that this specification for pH was not aligned with the raw material supplier specification of 7.0–9.5. Historically, the pH (average of 8.4) comfortably met the supplier specification but was close to the in-house specification 8.2–10.0. This was a supply risk due to the high probability of failing pH testing upon receipt.
A technical evaluation was performed to evaluate the impact of the pH attribute on the process performance and product quality. Because a titration step was added to the preparation of the sodium deoxycholate solution during DS manufacturing, it was recommended to remove pH from the sodium deoxycholate specification. This change improves the robustness of sodium deoxycholate supply with no impact on the DS manufacturing process or product quality.
At the time of assessment, removal of the pH specification required prior approval in Australia and New Zealand; was reportable with no restrictions in the US, Canada, EU, Great Britain, and Switzerland; and was not reportable in the rest of the world.
Case Study 4: Urea—Change from Noncompendial Pellets to USP Powder
Urea is typically the main component in the oxidation buffer in a DS process. The supplier discontinued urea in pellet form, which required a transition to USP compendial-grade powder (from the same supplier). This resulted in a raw material specification change in which all of the specifications for the pellets were included for the powder with the same limits (except appearance) and additional tests were added to comply with the USP monograph. Buffer preparation using urea powder was evaluated, and it was determined there was no impact on dissolution, pH, or conductivity parameters. However, because the pellet form was filed with the appearance of “small colorless or white pellets,” the change to powder required submission and approval of a variation by several health authorities before it could be implemented. Prior approval was required in EU, Great Britain, Australia, Switzerland, Turkey, and Israel, whereas notifications were submitted to the US, Canada, Brazil, Gulf Coast Cooperative, Egypt, and Colombia. The remaining countries considered the change as not reportable. The wide range in filing categories worldwide delayed global approval and implementation to manufacturing, which in a worst-case scenario could cause restrictions on supply.
| Description | Sodium deoxycholate | |
| Intended function | A mild detergent in a DS manufacturing process to remove host cell impurities such as lipids, nucleic acids, contaminating proteins, and pyrogens. |
|
| Required characteristics to perform the intended function |
Be present and soluble in insufficient quantity to achieve function. Sodium salt is highly soluble compared to free acid. |
|
| Material attribute | Target ranges | Justification/control strategy |
| Appearance | White crystalline powder | Basic GMP requirement tested for each batch–confirms correct material received and may be indicative of impurities present. |
| Identification | Pass/conforms | Basic GMP requirement. Raman or infrared tested for each batch–confirms correct material received and may be indicative of impurities present. |
| Loss on drying | ≤ 5.0% | No impact |
| Assay | ≤ 99.0% | No impact |
| pH of solution | control was implemented as part of the DS manufacturing process instead of at raw material release. A titration step with sodium hydroxide during the 10% solution preparation ensures the target pH is achieved regardless of the raw material pH ensuring no precipitation prior to manufacturing. The 10% solution is prepared and released as an in-process control based on a pH specification of 8.2 to 10.0. |
|
In summary, if raw material attributes are not critical, they should not be included in the specification, because changing or removing filed specifications can take months to years, making supply more challenging to manage. Also, attributes that have high variability have an increased risk of testing failures and risk to supply. Because not all attributes with high variability are deemed critical, a risk-based approach to testing should be taken to avoid risk to supply. Therefore, it is important to identify the critical attributes early during development and mitigate any risks upfront. Defining the raw material target attribute profile could enable the identification of ECs and submission categories when using ICH Q12 principles. For example, in the case of sodium deoxycholate, the pH of the solution may have been considered an EC because it is critical to ensure material solubility and the ability to perform its function. However, through the control of pH in the DS manufacturing process, the sodium deoxycholate pH attribute is not actually critical and was determined to have no impact on product quality.
Conclusion
As mentioned, it is critical to have a reliable supply of raw material to maintain robust drug supply in order to serve patients. Because of short-age-related challenges, implementing a global regulatory infrastructure is increasingly needed, specifically an infrastructure that is both flexible and predictable to provide more agility to react efficiently without product delays. Leveraging ICH Q12 principles such as ECs can streamline the number of postapproval submissions. In the future, more innovative regulatory approaches, as well as supply chain approaches to manage raw materials, could be envisioned. The use of structured content and data management in CMC regulatory submissions could potentially provide a direct link to proactively manage risks in the supply chain and communicate with regulators.11
In addition, employing the idea of quality management maturity to evaluate raw material manufacturing sites could perhaps enable an FDA rating system based on supplier excellence.12 Ideally, a sponsor could gain some regulatory flexibility if they were to switch suppliers to one that had an “excellent” rating. Finally, through convergence and reliance, a collaboration between regulators and industry stakeholders to develop and implement harmonized guidelines for raw materials could address multiple reviews of the same material and ensure that both regulator and industry resources are dedicated to only the most critical issues, ensuring uninterrupted supply of medicines to patients worldwide.
Reader note: This article was originally submitted to Pharmaceutical Engineering® in January 2022.
About the Authors



Acknowledgements
The authors acknowledge Jette Wypych, Mike Abernathy, and Satoshi Nagayama for their helpful discussions in the subject matter discussed in this article.


