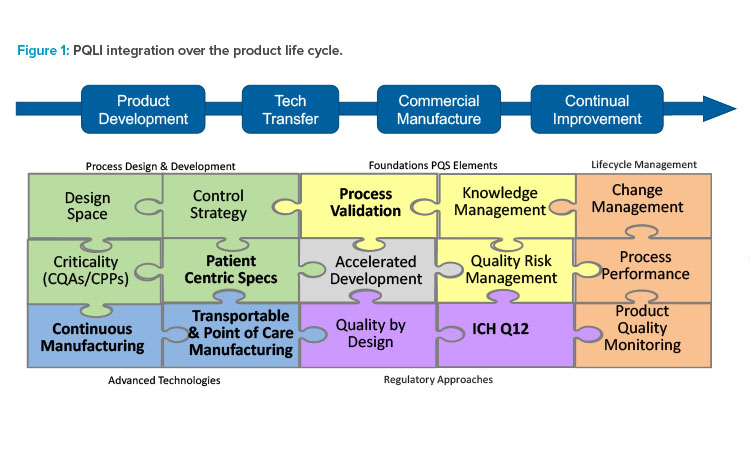
Implementation of continuous manufacturing introduces some unique challenges related to manufacturing process controls and assurance of product quality, especially for multinational companies that are trying to obtain global regulatory approvals without the benefit of harmonized regulatory guidance documents. Addressing these challenges has been and still is the focus of the CM team within PQLI.
As with any new technology, it is important that early adopters have opportunities to share their successes as well as pain points to help expand use of the technology and close gaps. The PQLI continuous manufacturing team has addressed these issues through biennial ISPE continuous manufacturing workshops. The inaugural continuous manufacturing workshop in 2016 focused on the business benefits and regulatory and technical challenges of the newly emerging technology. This workshop led to the initiation of the PQLI continuous manufacturing team. In 2018, the focus was on small molecule API and drug product, which was the more mature platform for continuous manufacturing at that time, with multiple products having gained regulatory approvals. The workshop brought together regulators, industry members, and academics from across the globe. Building on the success of the 2018 workshop, a virtual ISPE Continuous Manufacturing Workshop was held in 2020. As a sign of the maturation and expanded use of the technology, the workshop featured two parallel tracks for small molecules and biologics.
To ensure a broader reach beyond workshops, the team has also focused on publishing papers to address novel elements of continuous manufacturing. Two theme issues of Pharmaceutical Engineering® have been published. In May-June 2019, the focus was on holistic control strategy, process validation, sampling, and regulatory progress globally. The current issue focuses on the progress in the past two years, highlighting the advancement of continuous manufacturing for biologics, the global acceptance of continuous manufacturing, development and validation of a continuous manufacturing process, and summary of the work that is being done within ISPE in support of continuous manufacturing equipment development.
With the progression of ICH Q13 Continuous Manufacturing of Drug Substance and Drug Products, there will be opportunities for the PQLI continuous manufacturing Working Group to comment on the draft guidance document and to generate working products to support the successful implementation of the guide-line across our industry and around the world.
PQLI Transportable and Point of Care (POC) Working Group
Transportable and point of care manufacturing is the subject of the newest PQLI technical team; it is emerging as an enabler for speed to market by providing decentralized manufacturing and distribution closer to the patient. See Table 1 for different types of mobile manufacturing and the applications they support.
Process intensification creates opportunities for facilities with a smaller footprint that can be replicated cost-effectively, reducing supply risk through redundancy and allowing flexibility to respond to changes in market demand. Mobile manufacturing affords the opportunity to change the regulatory pathway in a science- and risk-based manner. The potential for portable manufacturing is immense with possible manufacturing applications for small molecule, biotechnology, and cell and gene therapy products, especially if utilizing smaller equipment such as with continuous manufacturing. Regulatory flexibility and speed can be achieved with strong collaboration between industry and regulators, as shown during the COVID-19 pandemic. The question for mobile manufacturing then becomes: can elements from this experience become part of the normal business process?
Risks to product quality for transportable manufacturing tend to be lower than traditional site changes due to the sameness or similarity of equipment and scale. Two scenarios are envisioned that avoid traditional scale-up and technology transfer issues: (a) the same transportable unit can be relocated from one location to another, and (b) the transportable unit can be replicated or cloned using the same design and equipment. However, in many regions the regulatory framework is not in place to provide benefit from the lowered risk related to transportable manufacturing. The lack of regulatory framework leads to unique challenges, especially for multinational companies that are trying to obtain global regulatory approvals without the benefit of harmonized regulatory guidance documents. Addressing these opportunities and challenges is the focus of the Transportable and POC team.
The adoption of new technology must start with a level of understanding of the benefits and challenges that exist with implementation. To that end, the team will introduce the different types of mobile manufacturing and drive an understanding of the technical, quality, and regulatory aspects of the technology at various forums.
Table 1: Types of modular, mobile, and point of care manufacturing configurations.
| Manufacturing Type |
Volume Supported |
Applications |
| Portable on-demand modular cleanrooms (PODs) |
Low to moderate |
Clinical
Commercial
Lab |
| Manufacturing trailers |
Low |
Clinical
Lab
Some commercial |
| Portable skid |
Low to moderate |
Clinical
Commercial |
| Suitcase manufacturing |
Single dose |
Clinical
Commercial |
PQLI ICH Q12 Working Group
ISPE PQLI ICH Q12 Working Group was formed in 2019 with the focus on supporting the adoption of ICH Q12 guideline on pharmaceutical product life cycle management by regulatory agencies and enabling the implementation of the ICH Q12 principles by pharmaceutical and biopharmaceutical companies. ICH Q12 is a transformational guideline that has a wide scope of applicability. ICH Q12 builds on the framework laid down in ICH Q8–Q11 guidelines and has the potential to remediate key remaining technical and regulatory hurdles that prevented the full adoption and implementation of flexible science- and risk-based approaches to postapproval CMC change management.
The progress to date on implementation of ICH Q12 has been slow. ICH is currently completing the preparation of training materials and is planning the initiation of a training program in fall 2021. The EMA published a note on EU implementation of ICH Q12 in March 2020, while the FDA was expected to issue its ICH Q12 implementation guidance in May 2021. Many other global health authorities such as Japan, Brazil, Canada, and China have discussed similar plans for issuing guidance for implementation of ICH Q12 in the next year or two.
Among the ISPE PQLI ICH Q12 Working Group’s recent accomplishments was a feature article in ISPE’s Pharmaceutical Engineering May-June 2020 issue that discussed key aspects of ICH Q12 and highlighted major changes from Step 2 (draft) to Step 4 (final) guideline document. The working group has also played a leading role in organizing ICH Q12–focused sessions at conferences and workshops featuring a diverse group of speakers from both the biopharmaceutical industry and regulatory agencies at the 2020 ISPE Biopharmaceutical Manufacturing Conference, 2020 ISPE Annual Meeting & Expo, and 2021 FDA-Xavier PharmaLink conference. Some of the key themes that emerged from these meetings included the importance of robust criticality assessments and the effective communication of their results and data to regulators in support of proposed established conditions (ECs); a wide variety of terminology and approaches used by the applicants and the need for greater alignment in this area; and a better understanding of what constitutes a robust pharmaceutical quality system and how it should be conveyed to regulators. Significant interest arose around key lessons learned from the recent FDA Pilot on Established Conditions and how the FDA plans to implement ECs and product life cycle management (PLCM) documentation in the future. Some of these questions and concerns have been addressed in the recently published FDA draft guidance on ICH Q12 implementation.
Going forward, the working group plans to continue its efforts to drive global ICH Q12 adoption, define key opportunities and challenges with Q12 implementation, and develop examples and trainings that help improve the understanding of the key guideline principles. ICH Q12 is a complex guide-line with novel tools and approaches, some of which are currently not fully compatible with the existing legal frameworks in certain markets. Therefore, for the full potential of ICH Q12 to be realized, it is critical to foster a robust global dialogue around current implementation challenges and to share the best implementation practices. To achieve its ambitious mission, the working group seeks to expand its partnerships with other cross-industry groups, including those under ISPE PQLI initiative, as well as ICH and other global regulatory bodies.
PQLI Patient Centric Specifications Working Group
Patient-centric specifications are those focused on delivering a quality product based on patient needs. Historically, regulatory agencies evaluated a marketing application’s proposed manufacturing, drug substance, and drug product specifications by relying on the developmental work and the clinical batch history. The challenge is that often there are only a few clinical lots, especially if the marketing application was submitted under an accelerated developmental program. Limiting the evaluation criteria to the clinical batches does not take into consideration the process understanding or prior knowledge from similar products, animal studies, publications, and process controls/attributes risk that are also relevant to the product quality.
In the past three years, this PQLI team has been involved with sharing the patient-centric specifications topic in different conferences and forums. The feedback from the presentations has been very positive from the pharmaceutical community and the regulatory attendees. Pharmaceutical company representatives acknowledge that they have all faced the same challenges to obtain a globally accepted specification approach, which has resulted in complexity within the pharmaceutical companies’ regulatory and operations groups to support the divergent marketing application details. The FDA has also acknowledged that a patient-centric specification approach is acceptable. In fact, the FDA has created an internal procedure document for their assessors to drive a more consistent approach in determining approvability of specifications and controls based on all of the relevant data and not just on the manufacturing history However, the team fully recognizes that to be successful, patient-centric approaches need to be adopted worldwide.
As the team looks to continue the conversations about this topic, they are focused on three goals. The first goal is to host an ISPE webinar to broaden exposure to the topic to ex-US regulatory agencies. An FDA representative will also be among the speakers to provide the regulators’ perspective. Second, the team plans to publish a discussion paper in support of changing the ICH Q6A/B documents’ language to be consistent with the patient-centric specification approach. Part of the confusion about appropriate specifications can be attributed to the current language in both ICH Q6A and 6B,, which are now over 20 years old. Some of the text is conflicting even within the guidance documents when viewed from a patient-centric perspective. In contrast, more recent ICH guidelines, such as ICH M7 on mutagenic impurities and ICH M9 on biowaivers, have elements that are consistent with patient-centric specification setting. Finally, the team is looking to continue to advance the topic through publications. The PQLI Working Group published a paper for small molecule considerations of patient-centric quality standards and is working on a paper specific to biologic specification setting. Even though new biological entities are more complex than their small molecule new chemical entities, there are still opportunities to support a patient-centric approach in the biologics marketing applications.
PQLI Process Validation Working Group
The Process Validation (PV) Working Group is the most mature of the PQLI teams; it was founded about 10 years ago and remains very active. The process validation team is focused on the practical implementation of life cycle approaches to process validation. Because of its broad nature, process validation has natural linkages to many other PQLI and ISPE topics under development.
Initially, the team was focused on understanding challenges in practical implementation of process validation to the product life cycle arising out of the regulatory guidance such as the FDA’s 2011 Process Validation guide. Questions addressed included use of statistics, implementation of control strategy, planning and management of continued/ongoing process verification, application of process validation to legacy products, and techniques to determine the number of batched required for process performance qualification (PPQ).
The process validation team has been active in presenting best practices from ISPE member organizations, hosting work groups and discussions on challenging topics, and disseminating information through published articles and presentations. The team has published nine discussion papers over the past decade covering a wide range of process validation-related topics, including “Stage 3 Process Validation: Applying Continued Process Verification Expectations”; “Lifecycle Approach to Biotech Process Validation”; “Implementing Lifecycle Validation Practices at Contract Manufacturing Organizations”; “Determining Number of Process Performance Qualification Batches Using Statistical Tools”; and “Process Validation in Context of Small Molecule DS and DP Continuous Manufacturing Processes.” These discussion papers are provided free to ISPE members.
The process validation team regularly holds sessions at the ISPE Annual Meeting on process validation-related topics. Additionally, the team has conducted five well-attended stand-alone process validation workshops between 2012 and 2019, where both industry SMEs and regulators shared their experience on this subject. Despite process validation being a more mature topic, there is still enormous demand for some of the original concepts developed by the team, especially in a webinar format. Members of the process validation team developed ISPE’s Process Validation Training course and have contributed to many regulatory and affiliate conferences around the world. ISPE instructors have also provided multiple trainings to regulators worldwide on process validation topics.
In 2019, a subgroup of the process validation team authored ISPE’s Process Validation Good Practice Guide. This guide built on the work of the team and contains practical guidance covering all three stages of the process validation life cycle as it is interpreted worldwide. It discusses process validation in the US and EU and in emerging markets such as Asia Pacific and other parts of the world. The guide contains six detailed appendices covering case studies and application of statistical techniques to process validation.
The process validation team is still very active, with 50 members that meet monthly. The team continues to work on evolving new process validation content and delivering knowledge. Topics of current interest include applying science- and risk-based thinking to emerging process validation challenges for new modalities such as cell and gene therapy, products under accelerated development, and continuous verification.
Conclusion
Although much has changed since PQLI was initiated, one constant is the ability of PQLI technical teams to advance new regulatory approaches based on current science and technology. The life cycle aspect of PQLI covers not only pharmaceutical product life cycle, but also the life cycle for the technical teams themselves. New topics are added to PQLI as they emerge and sunsetted as they mature into standard practice. The chairs of PQLI and the team leads welcome ideas for new topics and nominations of experts who are ISPE members and willing to contribute to technical teams. ISPE members with deep subject matter expertise should contact Regulatory@ISPE.org if interested in participating in PQLI teams as contributors.