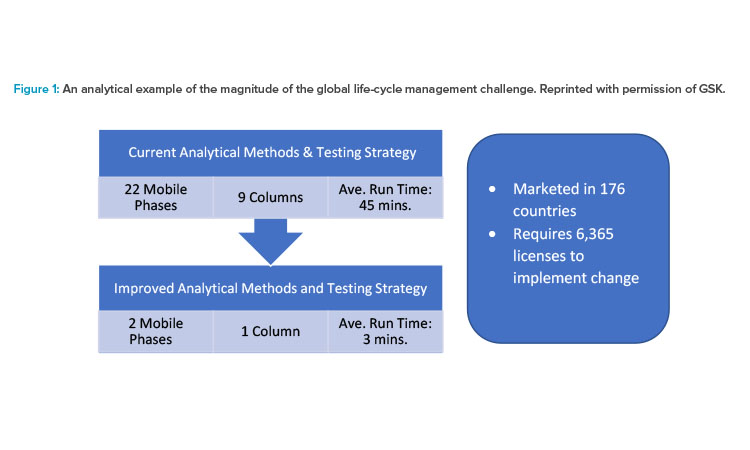
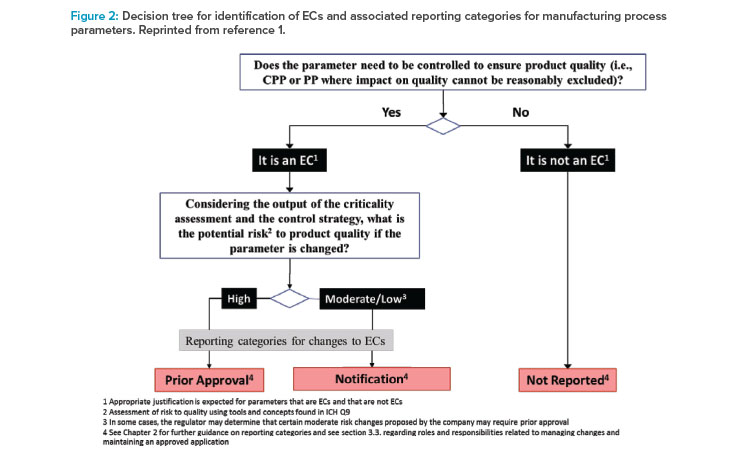
During the Step 2 draft review and consultations, it became evident that the three-tiered approach to classify the criticality of process parameters has been adopted throughout the industry. However, the proposed definition for key process parameter introduced the concept of “process consistency,” which is not defined in any ICH guidelines. In addition, fundamental provisions of the ICH Q12 guideline indicate that the management of changes impacting process consistency that do not directly impact quality should be managed under the market authorization holder’s Pharmaceutical Quality System (PQS). Consequently, the final version of ICH Q12 retained the overall three-tiered approach to process parameter classification (Figure 2), but the reference to key process parameter was replaced with “other process parameters where an impact on product quality cannot be reasonably excluded.” As a result, ICH Q12 defined more clearly the scope for the assessment of Established Conditions relative to their impact on product quality. Furthermore, regardless of the approach used, manufacturing process descriptions in Module 3 are expected to remain suitably detailed and to include both Established Conditions and supportive information. These changes in the final version mirror the key provisions from the 2017 EMA guideline on manufacture of the finished dosage form.
In the final version of ICH Q12, the discussion of the different approaches available to identify Established Conditions in the manufacturing process was reworked and enhanced to improve clarity. Minimal and enhanced approaches were grouped together as parameter-based approaches because they are primarily focused on the Established Conditions related to process inputs. The performance-based approach was set apart to highlight its primary focus on the Established Conditions related to process outputs. The performance-based approach description was clarified with some additional illustrative examples of the approach’s scope of applicability, such as “in-line monitoring of relevant attributes or with feedback controls or optimisation algorithms to achieve the relevant targets for that process step.”
Product Life-Cycle Management
The Product Lifecycle Management document was one of the key tools introduced in the Step 2 draft guideline. It is intended to serve as central repository for Established Conditions and reporting categories and also includes a summary of the product control strategy, Post-approval Change Management Protocols, and postapproval CMC commitments, as applicable. The Product Lifecycle Management was envisioned to contain a summary of key elements of the product control strategy that justify and explain the selection of Established Conditions. However, in the final version, the summary of the product control strategy was removed to simplify the Product Lifecycle Management document and make it applicable across all ICH regions. In the Step 2 draft, the location for the Product Lifecycle Management document was conspicuously absent, in deference to existing regional/local regulatory requirements. Following critical feedback received during the draft review, the final version of ICH Q12 clarified that the Product Lifecycle Management document should be placed in 3.2.R or for some regions in Module 1 (e.g., Japan). Finally, the final version of the guideline emphasized that submission of the Product Lifecycle Management document is “critical” when a market authorization holder wishes to use any of the ICH Q12 risk-based approaches to define and propose their own Established Conditions with associated reporting categories.
Postapproval Changes
The chapter on postapproval changes to authorized products was expanded in the final version of ICH Q12 to describe key considerations for structured approaches for some of the more frequent CMC changes, such as analytical method improvements, manufacturing process scale changes and improvements, and alternative packaging components. The detailed example of an approach for analytical procedure changes was moved to Annex II to allow flexibility in updating the example as appropriate. The ICH Q12 Expert Working Group noted, “The flexibility provided in Annex II may not be available in all regions and in all situations; some specific changes may require prior approval as defined in regional guidance.” Specific guidance on the type and amount of stability data expected to support postapproval changes was given its own chapter.
Post-approval Change Management Protocol is a regulatory tool providing prospective transparency and expectations for regulatory reporting categories for specific postapproval changes. Post-approval Change Management Protocol has been an integral ICH Q12 concept throughout the guideline’s development and must be approved by regulatory authorities. With the implementation of ICH Q12, Post-approval Change Management Protocol can help ensure robust change management globally by providing a consistent approach to regulatory reporting categories across ICH regions.
ICH Q12 Annex
Between Step 2 and Step 4, the ICH Q12 Annex documents were also revised and updated. The examples provided in Annexes IA and IB on how to identify Established Conditions in the manufacturing processes of chemical and biological products were reduced to focus on fewer individual process steps. However, the justifications substantiating these examples were significantly expanded to provide increased understanding and practical implementation of these concepts across all three risk-based approaches provided in the guideline. Annex IC was added to specifically illustrate how Established Conditions can be identified for analytical procedures, using as an example capillary electrophoresis for a biological drug substance under minimal development approach. The examples of Post-approval Change Management Protocols for chemical and biological products were moved into Annexes ID and IE, respectively, but otherwise remained largely unchanged. An example of a Product Lifecycle Management document, with a few important clarifications based on public comments, was moved into Annex IF. The revised example is not an exhaustive list of all Established Conditions that may be included in an application; it serves only as an illustrative example for manufacturing process-related Established Conditions for a small molecule drug. A comprehensive list of all Common Technical Document (CTD) sections containing Established Conditions is provided in Appendix 1 of the main guideline.
As mentioned previously, Annex II of the final guideline contains the detailed example of a structured approach to analytical procedure changes.
Conclusion
The adoption and implementation of ICH Q12 principles is expected to transform the global regulatory environment by leveraging the risk- and science-based concepts of Quality by Design articulated in ICH Q8, Q9, Q10, and Q11. ICH Q12 should drive regulatory convergence for postapproval changes and enable continual process improvement and the adoption of innovative technologies that increase quality assurance and reduce the volume, time, and resources needed to prosecute regulatory applications. The use of Established Conditions provides clarity and improves transparency between regulatory authorities and industry. Post-approval Change Management Protocol should improve clarity for verification, documentation, reporting, classification, and implementation of CMC postapproval changes. ICH Q12 encourages the industry to embrace the new paradigm to prospectively consider and assess process and product improvements and innovations by using the new tools and approaches to expedite regulatory assessments and inspections.
The implementation of ICH Q12 may warrant increased interactions between industry and regulatory agencies in the short term. However, the long-term expectation is that use of ICH Q12 will decrease the need for postapproval filings and agency interactions. The initial interactions may take many forms, ranging from product-specific meetings to open forums. ICH intends to develop additional training materials, which should be beneficial to both the industry and regulators.
Appendix: Detailed Table of ICH Q12
An appendix to this article comparing the Step 2 draft of ICH Q12 and the final guideline is available.
Download Appendix