Good Engineering Practice in Risk-Based Commissioning & Qualification
Over the years, the roles and responsibilities of engineering and quality/validation personnel for commissioning and qualification (C&Q) activities have evolved. Now more than ever, commissioning and qualification approaches based on quality risk management (QRM) principles rely heavily on engineering and the application of Good Engineering Practice (GEP) to provide documentation for the qualification package.
This article explores the current industry transition of principle ownership of process manufacturing performance verification from quality/validation to engineering—a transition that empowers both engineering and quality personnel with the tools to deliver true quality by design, resulting in improved product quality, improved patient safety, and increased speed to market.
The Evolution of QRM as the Basis for Commissioning and Qualification
The ISPE GAMP Guide introduced the V Model in 1994.1 In 2001, the first edition of ISPE Baseline Guide, Vol. 5, commissioning and qualification (the “Commissioning and Qualification Guide”) introduced commissioning and the use of impact assessments as a formal means to identify a system’s potential impact on product quality.2 This commissioning and qualification process became the pharmaceutical industry’s standard practice, but manufacturers have rarely implemented it to its full potential with respect to efficiency or the integration of engineering and quality roles and responsibilities.
After the first edition of the commissioning and qualification Guide was published, additional resources became available. ICH Q8, Pharmaceutical Development, introduced and defined the concepts of critical quality attributes (CQAs) and critical process parameters (CPPs); ICH Q9, Quality Risk Management, provided a framework for a holistic quality risk management program; and ISPE published a white paper, Risk-Based Qualification for the 21st Century, describing the application of quality risk management principles to commissioning and qualification.3, 4, 5 ASTM E2500, Specification, Design, and Verification of Pharma/Biopharma Manufacturing Systems and Equipment, provided guidance on how these concepts could be integrated into a science- and risk-based approach to commissioning and qualification,6 and ISPE published two additional guides—one describing the ASTM E2500 approach in detail, and one describing a transitional approach.7,8 These industry efforts culminated in the 2019 publication of the second edition of the Commissioning and Qualification Guide,9 and there are high expectations that this edition will establish quality risk management-based integrated commissioning and qualification as the industry-standard approach, using common terminology and methodology. (The other ISPE guides described here are no longer available.)
Engineering vs. Quality Roles in the QRM-Based Commissioning and Qualification Model
As defined and standardized in the second edition of the commissioning and qualification Guide, commissioning and qualification is an integrated process for establishing that systems are suitable for their intended purpose. Within this commissioning and qualification process:
- Commissioning is the engineering process for delivering systems that meet established design requirements and stakeholder expectations.
- Qualification is the quality process for demonstrating and documenting that critical systems are suitable for their intended purpose.
- Verification is any activity that supports those processes and demonstrates that systems are suitable for their intended purpose.
Engineering subject matter experts (SMEs) play an integral role under this model. Process development and technical operations SMEs develop, define, and ensure the technical transfer of product and process knowledge (PPK) during process development. Engineering is responsible for:
- Ensuring that product and process knowledge is incorporated into the user requirements specification (URS)
- Identifying user requirements that impact critical quality attributes and critical process parameters and that contribute to process and system risk assessments
- Completing design development
- Performing design review
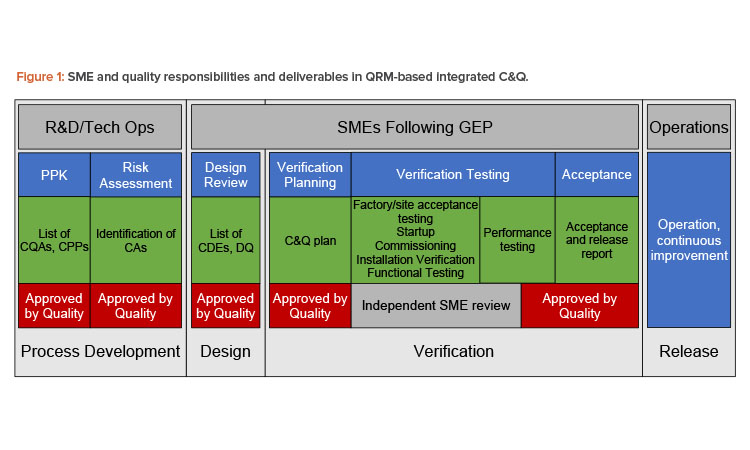
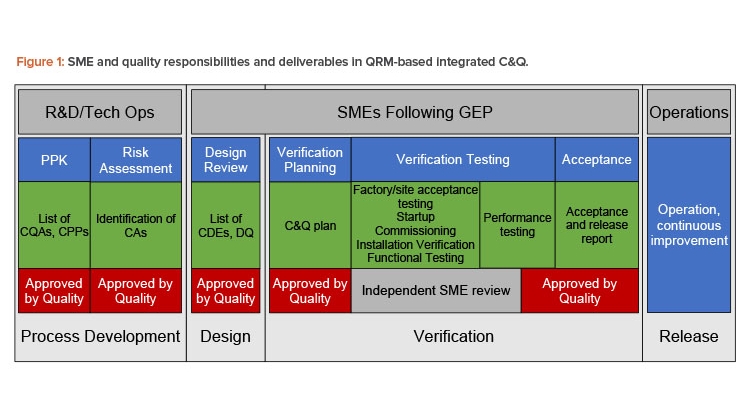
In sum, engineering is responsible for implementing the risk control strategy—that is, ensuring that critical design elements (CDEs) of the system identified through risk assessment are satisfied by the design. As a result, engineering is a key contributor to development of the design qualification (DQ) based on the final approved design. During commissioning and qualification, engineering SMEs develop the commissioning and qualification plan; perform vendor assessments where they could be beneficial; develop, approve, execute, and review verification testing; and manage discrepancies and changes. Engineering personnel/SMEs then develop the acceptance and release reports.
Under this model, quality personnel have an oversight role with the following responsibilities:
- Ensuring that the commissioning and qualification process uses product and process knowledge
- Ensuring that the critical quality attributes and process parameters are incorporated into the user requirements specification
- Approving the risk assessment, including the identification of critical aspects (CAs) and the acceptability of residual risk
- Approving the identification of critical design elements and their acceptance criteria
- Ensuring that testing and documentation are commensurate with risk by approving the commissioning and qualification plan, vendor assessments, verification testing deviations involving critical aspects and design elements, and acceptance and release reports
In Figure 1, the responsibility of engineering is apparent. Engineering is responsible for delivering systems fit for the intended purpose, including developing, executing, reviewing, and approving all testing and documentation required to deliver those systems and documenting their fitness for purpose. This model does not differentiate testing for commissioning and qualification testing from other testing; all testing is verification—that is, all testing contributes to demonstrating and documenting that systems are fit for their purpose.
The benefit of this approach to quality is significant. Once critical aspects and design elements are documented with acceptance criteria in the design qualification, quality personnel can narrow their focus to their most important priority: process risk mitigation that ensures product quality and patient safety. Table 1 summarizes roles, focus, and responsibilities for engineering and quality in quality risk management-based commissioning and qualification.
| Engineering | Quality | |
|---|---|---|
| Role | System delivery | Product quality and compliance |
| Focus | Fitness for intended use | Ensuring that risks to product quality (patient safety) are identified, and adequate controls are proposed to provide an acceptable risk level Ensuring that the designed controls are tested, have been installed, and operate to meet the specifications and support qualification |
| Responsibilities | Development of URS, design development, design review, installation, verification of the installation and operation vs. the design specifications, engineering change management (ECM), commissioning summary report (testing and documentation) |
Approval of the design qualification, test strategy, and acceptance criteria Approval of engineering change management for direct-impact systems Approval of the equipment qualification and releasing the system to the next stage |
Current Industry Practice
There is a considerable gap between the principles set forth in the second edition of the commissioning and qualification Guide and current industry practice. Prior to publication of the first edition of the commissioning and qualification Guide, industry had not adopted a structured approach to commissioning. Presently, companies typically have no or limited commissioning procedures. Therefore, systems could be “qualified” but would not truly be ready for operations; additional start-up, debug, and engineering runs would be required for a fully functioning process because companies lacked user requirements specification, design review, and ade-quate understanding of critical aspects.
The first edition of the commissioning and qualification Guide presented a model in which a system underwent impact assessment and then was designed, built, commissioned, and qualified.2 Under this model, systems were intended to be commissioned following good engineering practices.10 A system-level impact assessment (SLIA) determined which systems had impact on product quality (classifying systems as having direct, indirect, or no impact), and a component criticality assessment (CCA) determined whether components within a direct-impact system were critical or noncritical to product quality.
The system-level impact assessment and component criticality assessment were “bottom-up” (i.e., from the equipment/system to the product) forms of risk identification focused on the system, rather than on the product and product and process knowledge, which led to binary “yes/no” risk evaluation. In practice, segregating commissioning from qualification testing and introducing a “leveraging” mentality devalued the importance and contribution of good engineering practice to the commissioning and qualification process, leading to an inefficient process in which vendor testing (factory/site acceptance testing, etc.) was not considered part of the process. The first edition of the commissioning and qualification Guide proposed that engineering documentation be used or “leveraged” to support qualification.2
In practice, this approach reduced the redundant testing to qualification of critical components in direct-impact systems and evolved into the concept of “leveraging” commissioning testing into qualification testing. As a result, quality personnel required pre- and postapproval of any engineering testing that was intended to be leveraged. Increased documentation scrutiny was often seen as a hindrance to engineering, and engineering testing that was not intended to be leveraged was often viewed as non-value-added.
Even as firms move to adopt quality risk management-based integrated commissioning and qualification, most projects repeat testing in installation/operational qualification (IOQ). This approach is based on legacy practices and reflects the perception that all user requirements specification items designated as “quality” require a clean run test within installation/operational qualification. However, this approach diminishes the manufacturer’s ability to use value-added testing and drives up installation/operational qualification hours. A small subset of companies have successfully used an integrated commissioning and qualification process to limit installation/operational qualification to a summary report or gap assessment of commissioning with minimal repeat testing. In practice, good engineering practice is generally not viewed as an engineering quality process necessary to support quality and validation. To the contrary, good engineering practice may be followed as a matter of procedure or seen as a non-value-added hindrance to project progress. Additionally, many current commissioning and qualification approaches produce a mindset of “We’re only in the commissioning stage; systems will be verified during qualification.” As result, most engineering SMEs involved in commissioning focus their efforts on system aspects that do not impact product quality, which is the opposite of where engineering SME efforts should focus.
In general, good engineering practice systems are lacking in robustness, maturity, implementation, compliance, or all of these, and are therefore not suitable for under-pinning a quality risk management-based integrated commissioning and qualification approach. Furthermore, when the quality unit lacks trust in good engineering practice as an engineering quality system, it requires use of its own quality systems, such as quality change control in lieu of engineering change management (ECM), to provide appropriate oversight and control of all commissioning and qualification activities. The end result is additional, less-efficient, more costly processes and additional documentation, testing, and effort with no commensurate, additional reduction or mitigation of product quality risk.
However, capital project timelines are shrinking due to the accelerated approval process, Agile manufacturing, and pressure to shorten the time to market. Corporate engineering is challenged to deliver projects in record time. The typical phase-gate model is being replaced by Agile and value-engineering approaches, and there is much less tolerance for repeated testing and issue discovery during start-up, debug, and engineering runs.
The Cost of Poor (Engineering) Quality
So, what happens when engineering testing is considered insufficient for commissioning and qualification needs? The following are some real-world examples:
- Protocol development is driven by the turnover package (TOP) rather than by the risk control strategy (i.e., identified critical aspects and design elements). In one case, a turnover package deemed acceptable by engineering was considered unacceptable by the validation team. That team then took a “We cannot start protocol generation until the turnover package is ready” stance, resulting in delayed protocol generation that immediately affected the overall project schedule.
- Discrepancies that do not impact product quality increase overhead. Issues that have no impact on product quality, such as incorrect make or model number, maybe among the discrepancies observed during engineering verification testing. Documenting these issues requires multiple-page forms with multiple steps and signatures, but the ultimate conclusion is “As-installed meets requirements; update the specification.” Such quality oversight does little to resolve these discrepancies but adds considerable churn and time to the project.
- Execution schedules are delayed by unnecessary verification-activity dependencies. For example, performing drawing walkdowns and generating redlines during field walks months after receipt and installation can result in quality determining that operational qualification cannot start until installation qualification is complete. This causes delay while drawing review and updates are completed.
- Critical execution paths are driven by systems with no impact on product quality. Some stakeholders believe that functional or operational verification (traditionally called “operations quality”) cannot start until all predecessor systems are “qualified.” For example, a company may presume that all feed and intermediate systems in a pharmaceutical water train (municipal water supply, soft water, reverse osmosis water, water for injection) must be qualified as a prerequisite to qualifying a clean steam generator.
- Non-value-added redundant testing is done. For example, when continuity and loop checks performed by the electrical vendor lack appropriate documentation or are not approved by quality, the commissioning and qualification team repeats those checks. Similarly, if in-place loop calibration performed by instrument ser-vices through automation is not considered to be valid verification testing, the commissioning and qualification or automation team redoes the work.
Many assume that engineering’s role is to get the project done, and those working invalidation will find the problems and provide the documentation. However, this mindset cannot be maintained. Engineering must embrace the efficacy and efficiency gained through a robust good engineering practice-based engineering quality system to deliver facilities, utilities, equipment, and systems that are demonstrated and documented to be suitable for their intended purpose.
Building Trust between Engineering and Quality
Building trust between engineering and quality personnel may require both procedural and cultural change for those in engineering and a paradigm shift for those in quality. All stakeholders must regard good engineering practice systems to be a good business practice that ultimately delivers the project faster and with fewer issues; these systems are a critical component of quality risk management-based integrated commissioning and qualification.
Systems may need to be developed or matured—a spreadsheet on an engineer’s computer is insufficient for engineering change management. The suitability of good engineering practice systems to support and enable quality risk management-based integrated commissioning and qualification may need to be demonstrated through trials or pilot projects assessing engineering product quality. And the quality unit will need to change from a project quality control mindset to a project quality assurance mindset.
The good news is that, in most cases, both engineering and quality personnel welcome and desire this change. Engineering wants to deliver systems that meet requirements, and the quality unit wants to focus on product quality instead of having their efforts diluted by overseeing aspects of project delivery that do not impact product quality.
The Integration of QRM and GEP
To understand the importance of engineering SMEs and the application of good engineering practice, one must first understand how quality risk management impacts the commissioning and qualification process.
![QRM process overview. Reprinted from ICH Q9 [4, p.4]. © European Medicines Agency, 2015.](/sites/default/files/2020-04/0520_PE_MJ_TechBennet_02.jpg)
Background on the QRM Process
Quality risk management is a holistic process in which management policies, procedures, and practices are systematically applied to the tasks of analyzing, evaluating, and controlling risks (see Figure 2). Risk assessment, which is perhaps the most familiar part of the quality risk management process, includes the identification, assessment, and evaluation of risk to product quality and patient safety. Risk identification answers the question, “What could go wrong?” Risk control involves decision-making to reduce or accept the level of qualified or quantified risk. Risk reduction is the process of mitigating or avoiding identified risks by reducing severity, decreasing the probability of occurrence, or increasing the likelihood of detection. Applied to a manufacturing process, quality risk management results in the definition of a risk control strategy—the collective design controls, alarms, and procedural controls implemented to mitigate or avoid unacceptably high risk to product quality or patient safety.
Identifying risks to product quality and patient safety requires product and process knowledge, namely the understanding of critical quality attributes and process parameters. Risk to product quality is defined as a failure to meet a product’s critical quality attribute, and a process risk is defined as a failure to maintain a critical process parameter. Thus, product and process knowledge, as critical quality attributes and process parameters, is the input to risk assessment. Following the application of risk control, critical aspects, which collectively mitigate unacceptable risk to product quality, become the output of risk assessment (i.e., the risk control strategy).
| Good Engineering Practice Perspective | QRM Perspective |
|---|---|
|
|
As quality risk management is applied to process design, critical aspects are used to develop critical design elements (design features or functions of an engineered system that are necessary to consistently manufacture products with the desired quality). As quality risk management is applied to commissioning and qualification of a system, identification of critical design elements informs the verification strategy, such that these elements are tested commensurately with risk to product quality and patient safety, to verify that their installation and operation are fit for their intended purpose and that system critical aspects are met.
Good Engineering Practice Objectives
Good engineering practice, which includes elements of project management and project controls, ensures the following:
- Systems are specified, designed, and installed and operate in a manner that meets operational, maintenance, safety, health, environmental, ergonomic, industry, statutory, and regulatory requirements—including GxP requirements.
- Process risks to product quality are identified, assessed, and mitigated in system design, installation, and operation.
- Appropriate planning, specification, design, installation, verification, acceptance, and maintenance documentation is created throughout the system life cycle.
- Suitable oversight and control are provided for construction, installation, and execution verification activities.
In sum, good engineering practice systems—including design review, engineering change management, good documentation practice, document/drawing control, vendor qualification, construction quality, commissioning, issue/punchlist management, and asset management—comprise an engineering quality system that underpins the quality risk management-based, integrated commissioning and qualification process. Appropriate engineering SMEs define system requirements and specify, design, and verify the system in an efficient, effective, integrated approach. When a robust engineering quality system built on good engineering practice is implemented, the quality unit and its related quality systems—including quality change control, good documentation practice, document control, deviation management, corrective and preventive action, etc.—can properly focus on product quality and patient safety.
Let’s review some of those good engineering practice elements and their relationships to quality risk management
Design Review and Verification
According to ASTM E2500,6 design reviews are:
Planned and systematic reviews of specifications, design, and design development and continuous life-cycle of the manufacturing system. Design reviews evaluate deliverables against standards and requirements, identify problems, and propose required corrective actions.
The purpose of the design review can be considered from two perspectives: Good engineering practice and quality risk management (see Table 2).
The commissioning and qualification Guide (§5.3.3, Design Review Process) states:9
The effort, formality, and documentation of DRs [design reviews] should follow the ICH Q9 principles of being commensurate with the level of risk. DR documentation may take the form of engineering meeting minutes or notes. For highly critical systems, DRs may be more focused and detailed.
Per ASTM E2500,1 verification is:
A systematic approach to verify that manufacturing systems, acting singly or in combination, are fit for intended use, have been properly installed, and are operating correctly. This is an umbrella term that encompasses all types of approaches to assuring systems are fit for use such as qualification, commissioning and qualification, verification, system validation, or other.
As such, verification, like design review, can be considered from good engineering practice and quality risk management perspectives.
- From the good engineering practice perspective, verification includes fitness for intended use for all aspects of a system and encompasses both requirements that impact product quality (product/process user requirements) and requirements that do not (general user requirements).
- From the quality risk management perspective, verification includes fitness for intended use for critical aspects of a system and encompasses product/process user requirements only. As with design review, the scope of verification activities should be commensurate with the level of risk.
So, how does design review enable quality risk management-based, integrated commissioning and qualification? First, design review minimizes implementation of systems that fail verification testing by ensuring that the design satisfies user requirements prior to procurement or fabrication of the system. As a result, changes, redesigns, and rework during system commissioning are minimized.
Second, design review helps ensure that user requirements are well defined and appropriate. To perform verification, user requirements—and therefore the design aspects that satisfy those user requirements—must be specific, realistic, measurable, and verifiable. Verifying these aspects of the design through design review supports and facilitates verification of the system once implemented.
Third, and most importantly from the quality risk management perspective, design review supports definition of the process risk control strategy and identification of the system’s critical aspects. Design review identifies process risks to product quality posed by the design and determines the acceptability of those risks. Where identified risks are unacceptable, design review ensures that the design acceptably mitigates those risks. Thus, the design features that mitigate risk to product quality are identified as critical aspects, which, when combined with risk assessment, directly inform the verification strategy to ensure that testing is commensurate with risk.
Engineering Change Management
Engineering change management is an established engineering procedure for managing proposed changes, including request, impact assessment, implementation planning and execution, implementation verification, documentation, request closure, and follow-up. It is a system life-cycle process that begins at system definition and continues through design, verification, operation, and decommissioning. Engineering change management ensures that, through defined processes, proposed changes are assessed for project or operational risk, assessed for impact on product quality and patient safety, and implemented in a manner that is managed, tested, and documented commensurately with assessed risk.
From the good engineering practice perspective, engineering change management offers a change-implementation process that is scaled to risk, complexity, and system life-cycle change, ensuring controls are appropriate to the assessed risk. Prior to system acceptance, proposed changes to project requirements or design specifications are assessed for impact to project scope, cost, and schedule. After system acceptance, proposed changes to operational assets are assessed for their impact on operation and maintenance.
From the quality risk management perspective, engineering change management ensures that quality oversight for direct-impact systems is applied to changes affecting critical aspects that are pro-posed after the design has been accepted through design qualification. After the system is qualified, subsequent changes are managed through the quality (or operational) change control system.
Engineering change management enables quality risk management-based integrated commissioning and qualification in several ways. Engineering change management ensures that the impact of proposed changes on system requirements is identified, assessed, and controlled. During verification activities, engineering change management ensures that testing and documentation related to implementation of proposed changes are managed and controlled. Engineering change management supports quality risk management for direct-impact systems by ensuring that risks to product quality related to proposed changes are identified, assessed, evaluated, and controlled to an acceptable level; that quality oversight of proposed changes is appropriate for the system life-cycle stage; and that implementation of proposed changes is tested in a manner commensurate with risk to product quality and patient safety. Engineering change management enables quality oversight to focus on changes to the system’s critical aspects and design elements that have the potential to affect product critical quality attributes.
Engineering Quality Process
An engineering quality process (EQP) is a subset of the overall quality management system (QMS) that provides and addresses quality practices for commissioning and qualification. An engineering quality process is a philosophy rather than a collection of tools and templates, and, like the overall quality management system, it emphasizes product quality rather than regulatory compliance. The engineering quality process establishes or references the quality risk management process for commissioning and qualification; provides the basis for sustainable, consistent, cost-effective, and practical engineering processes for project and operational life-cycle management; and plays a significant role in providing the practices and controls within the facilities and equipment quality system, which is one of the six systems included in the US FDA Drug Manufacturing Inspection Compliance Program’s “Six System” inspection model.11
From the good engineering practice perspective, the engineering quality process does the following:
- Provides a systematic structure to define, develop, and implement efficient and streamlined engineering processes
- Delivers systems that are fit for intended purpose
- Enables the assurance of quality and compliance with policies, regulations, and standards
- Reflects the scope of the commissioning and qualification program and provides associated tools and templates
- Incorporates technical expertise and best practices
From the quality risk management perspective, the engineering quality process:
- Underpins efficient implementation of a science- and risk-based approach to facility start-up
- Enables science- and engineering-based decisions for commissioning and qualification testing
- Delivers systems with product quality risk sufficiently managed through application of engineering standards and practices
An engineering quality process emphasizes product quality rather than regulatory compliance.
An engineering quality process enables quality risk management-based integrated commissioning and qualification through documented activities during system life-cycle stages and supporting systems throughout the system life cycle. During design and procurement, engineering quality process manages vendor/supplier management. During construction/system implementation, engineering quality process manages construction quality, commissioning, and handover. During routine operation, engineering quality process manages asset management, calibration, and maintenance. Throughout the system life cycle, engineering quality process manages document and drawing control, issue and punchlist management, and engineering good documentation practice.
An engineering quality process ensures that product quality requirements are incorporated into system requirements. During specification and design, an engineering quality process ensures that design decisions and design review outcomes are traceable and can be evaluated. During verification, an engineering quality process ensures that testing activities and documentation are suitable for use as verification and are commensurate with risk. During system acceptance and release, an engineering quality process ensures that a system’s fitness for its intended use can be determined from engineering testing results.
Conclusion
Under the quality risk management model, all testing adds value as verification activities, testing is commensurate with product risk, good engineering practice and engineering SMEs are emphasized in the process, and quality is focused on the identification, mitigation, and control of risks to product quality, as well as verification of the process risk control strategy. Under this model, good engineering practice as an engineering quality process enables the commissioning and qualification process.
As a result, systems are designed and delivered to specification more reliably, with reduced costs and shorter schedules. Issues are resolved and changes are managed faster, more efficiently, and with greater cost efficiency. Verification testing activities are performed more efficiently and result in more robust, science- and risk-based documentation of fitness for intended use to produce products that meet quality requirements.
In partnership with ISPE, CAI is engaged in a long-term effort to develop industry benchmarking data to assess maturity of quality risk management-based integrated commissioning and qualification programs and to provide a roadmap for firms interested in following current industry best practices. Additional data will be collected and trended over time. CAI will continue to report on industry progress, and participating firms are provided access to the full data set.
Additional Resources
1. US Food and Drug Administration. Pharmaceutical Development for the 21st Century—A Risk-Based Approach. Silver Spring, MD: US Food and Drug Administration, 2004.
2. US Food and Drug Administration. Guidance for Industry, Process Validation: General Principles and Practices. Silver Spring, MD: US Food and Drug Administration, 2011.
3. International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. “ICH Guideline Q10 on Pharmaceutical Quality System.” European Medicines Agency website. Published 2008. https://www.ema.europa.eu/en/documents/scientific-guideline/international-conference-harmonisation-technical-requirements-registration-pharmaceuticals-human_en.pdf
4. European Commission. “EudraLex, Volume 4: EU Guidelines for GMPs for Medicinal Products for Human and Veterinary Use. Annex 15: Qualification and Validation.” Published 2015. https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-4/2015-10_annex15.pdf
About the Author



