Validation 4.0: Case Studies for Oral Solid Dose Manufacturing
Three case studies on Validation 4.0 demonstrate how quality by design (QbD) principles, when applied with digitization, can verify processes in scale-up and technology transfer, and why blend and content uniformity matter for tablet integrity.
This article provides background on Validation 4.0 and presents case studies on applying Validation 4.0 in oral solid dose (OSD) manufacture. The ISPE Validation 4.0 Special Interest Group (SIG), part of ISPE’s Pharma 4.0™ initiative, has written on the issue of Validation 4.0 previously in this magazine, first in “The History & Future of Validation” to provide history and overview of validation in the pharmaceutical industry and a definition of continued process verification and continuous process verification.1 The group also published “Laying the Foundation for Validation 4.0,”2 discussing how the topic of validation has been “a central obstacle to adopting new concepts for quality,” and has indirectly slowed the uptake of the very technologies that are required to bring pharmaceutical manufacturing into Industry 4.0.
History of Quality by Design
OSD manufacture is a multifaceted operation consisting of raw materials and unit operations that transform the raw materials into finished product. Traditional approaches to process development and validation involve the manufacture of validation batches to show that the combined unit operations and raw materials produce intermediates and finished products that meet a predefined set of efficacy and performance characteristics.
Major issues in the traditional approach to validation are the lack of information on representative sampling; the difficulty of real-time monitoring and control; and the snapshot, rather than continuous manner, in which validation is performed.
In 2011, the US FDA released an updated process validation guidance document3 and included the concept of “continued process verification” with routine monitoring of process parameters and trending of data to have a process that is capable of consistently delivering quality product. They referred to ASTM E2500-07, 4 which states that quality by design (QbD) concepts should be applied to ensure that critical aspects are designed into systems during the specification and design process. In this article, there is no distinction between the terms “continued process verification” and “continuous process verification” with regard to US and EU definitions.
Industry 4.0 as an Enabler of QbD
To verify the consistency of every batch in a sufficiently timely and accurate fashion, and to cope with inherent variabilities, some level of digitization is required. Information technology is not mandatory for QbD; however, the case studies show the capabilities of modern data systems for effective process understanding and control. Those that are increasingly available and accessible can improve product quality and efficiency and produce better medicines at lower manufacturing cost.
Industry 4.0, as a global manufacturing revolution, relies on collecting and using data in electronic formats to understand and improve process and product performance beyond legacy paper and manual methods.5 QbD has a similar philosophy and encourages manufacturers to start their validation efforts at product conception so that control strategies are built into the process, by design. This also ensures that when the process is moved, or scaled up to commercial manufacturing, the entire quality strategy is in place. Increasingly, the pharmaceutical industry is looking to digitalization as an enabler of QbD using data from all available sources. The following case studies and the capabilities described in this article are simply not practical without modern measurement sensors and data analysis tools.
In 2004, the US FDA published its process analytical technology (PAT) framework guidance to promote the use of innovative technologies for the collection of timely quality data that ensure the quality of product throughout the entire manufacturing process, from raw material dispensing to packaging.6 Together a combined QbD/PAT approach provides a solid platform for continuous verification and digitalization, particularly when linked to digital solutions and ways of working such as manufacturing execution systems (MES), supervisory control and data acquisition (SCADA) systems, and industrial databases that allow fast retrieval of data over multiple batches, months, raw materials, and lots. This knowledge management within systems can be used to optimize manufacturing and allow some flexibility within design constraints.
It has been 20 years since the FDA released “Pharmaceutical CGMPs for the 21st Century—A Risk-Based Approach: Final Report,” 7 and during that time, improvements in computing, data warehousing, sensor technology, and control systems now provide OSD manufacturers with the digital tools to address the validation requirements associated with continuous verification/validation and to move to Industry 4.0/Pharma 4.0™ and Validation 4.0 with confidence.
Validation 4.0 Is QbD-Centric
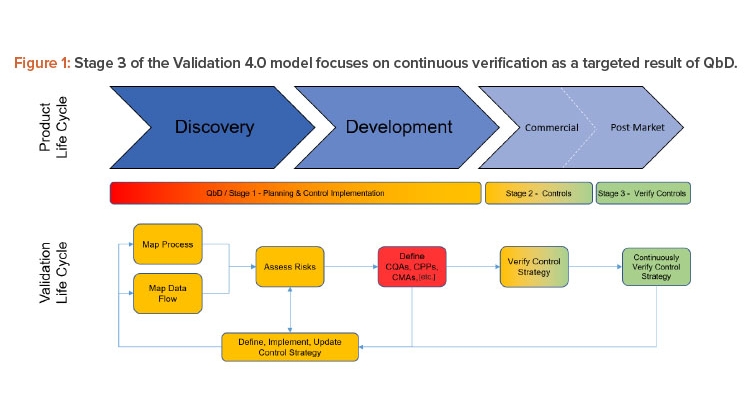
This article is focused on Stage 3 of the Validation 4.0 model (as shown in Figure 1). Verifying controls is done continuously with a frequency dependent on the ability to measure and identify risks from a process and systems perspective. One of the goals of Validation 4.0 is that Stage 2 ultimately absolves by moving directly to a state of continuous verification.
Traditional OSD manufacturing consists of a series of disjointed unit operations that transform raw materials and intermediates into finished products. Such processes are batch processes and transform large, bulk powders via the unit operations. OSD manufacture typically has a number of processes to deal with different raw material properties such as direct compression, roller compaction, and high shear granulation.
No matter which approach is taken, there is a common variable that is commonly overlooked in all three processes: raw materials. Raw materials are typically assessed using pharmacopeia monographs that focus on identification and purity aspects alone. The samples assessed are typically nonrepresentative and do not provide any information on how the materials will perform in the process.
QbD allows a manufacturer to build flexibility into manufacturing processes, rather than keeping the process fixed. If the process is fixed, inherent raw material variability cannot be accommodated by the process. This leads to substandard intermediates that, more often than not, result in issues in compression. Through improved sampling techniques and measurements from the initial stages of manufacturing, better understanding of the raw material variability allows the development of conformity or classification models. And technologies such as near-infrared (NIR) spectroscopy and powder characterization are used to understand the “process-ability” of materials.
In the QbD and Validation 4.0 world, digitalization tools and systems can use this information to adjust the process. Thus, QbD needs to assess all sources of variability, including inputs from outside the manufacturing floor, because variability from suppliers’ products can impact regulated customer’s processes. The ability to adjust a process is not unlimited and must be bound by certain rules that ensure quality. In QbD terminology, the boundaries around the process are known as the design space. By definition, the design space is established by design, i.e., through timely measurements that are linked to quality, or that are defined as critical quality attributes. The QbD approach has also introduced a new vocabulary associated with modern manufacture. Some of the important terms include the following:
- Quality target product profile (QTPP): Efficacy and performance characteristics that ensure the safety of the end user. Essentially, this defines the target product.
- Critical quality attributes (CQAs): Attributes of the product that ensure it is fit for its intended use when assessed against the QTPP. Essentially, these define the attributes that need to be met to meet the target product.
- Critical process parameters (CPPs): Controllable aspects of the manufacturing process that ensure the CQAs meet their defined targets. Essentially, these define what needs to be controlled so that the attributes will be met.
- Critical material attributes (CMAs): Attributes of the input materials that should be within appropriate limits or distribution to ensure the desired quality of the product.
Based on the previous points, validation becomes product- and process-centric through measurement data to maintain manufacturing within the design space, the resulting allowable limits of all the critical attributes and parameters that result in product that meets the QTPP requirements (also known as the desired state).
When a formulation is robust and the raw material characteristics are understood, the process can be adjusted within the design space, resulting in the highest-quality finished product. To fully achieve this and to incorporate it into an industrial production line, digitalization tools and appropriate sensor technology are required.
QbD allows a manufacturer to build flexibility into manufacturing processes, rather than keeping the process fixed.
An Introduction to Continuous Verification
A recent trend in pharmaceutical manufacturing is to move to continuous manufacturing (CM) systems. A CM system is a connected series of unit operations that converts small amounts of raw materials and intermediates at a time in small-scale equipment with much higher control and better sampling opportunities. The small amounts of materials processed at a time are called sub- or micro-batches. The combination of many sub-batches makes up a final batch, which is packed off while the next major batch starts and continues until the process is stopped.
In continuous manufacture, there is reduced opportunity to take physical samples from the process and assess them offline. When samples are removed, traceability becomes a problem, so timely information is required through highly digitized processing equipment and sensor technologies. In a CM system, every sub-batch is monitored and assessed for quality, thus providing a process chronology traceable back to unit doses in some cases. Such systems are not possible without QbD principles, PAT, and digitalization. In these types of systems, a real-time release strategy can be built into the process by design, allowing for materials to pass continuously through all unit operations to packaging and release without laboratory testing.
The principles of CM can also be applied to batch manufacturing operations if due consideration is given to better understanding of raw materials and correct process sampling strategies, based on large N sampling plans. To make large N sampling effective from an economical sense, the nondestructive, inline analysis of CQAs by PAT is required. In many cases, these PAT sensors will be NIR sensors and the data and models have to be taught by generating many spectra of the desired material. So there is a longer development phase when using PAT as a tradeoff to an improved process understanding and higher efficiency commercial production phase.
QbD and Continuous Verification
The basis for implementing QbD is described through the International Conference on Harmonisation of Technical Requirements for Pharmaceuticals for Human Use guidance documents Q8, Q9, and Q10;8 how they relate to ASTM;4 and the US FDA’s guidance and GMP principles.3, 6, 7
ICH Q8 establishes what is relevant for product development based on QbD principles and defines the concept of design space.9 The establishment of design space results from consideration of the risks to quality (ICH Q9), and the methodology used in these case studies starts with design of experiments and multivariate data analysis (MVDA) approaches to build predictive models based on available digital information from process equipment and process sensors. The iterative nature between Q8 and Q9 allows risk mitigation and the knowledge obtained is managed by the pharmaceutical quality system (PQS) of ICH Q10. 10
Extending the concept of the PQS in Validation 4.0 is knowledge management as a repository, where digitized information collected from real-time measurements is used in conjunction with process models. These models were developed during the early-stage development learning/design phases, which ensure the process is operating within the design space and thus producing product in its desired state.
Data systems for QbD aspects of Validation 4.0 include:
- Electronic process definition and control (i.e., integrated manufacturing execution system / electronic batch records for manual operations with under-lying industrial internet of things [IIoT], supervisory control and data acquisition systems, or distributed control systems for running automated equipment)
- Industrial databases that collect real-time data, alarms, and events (i.e., data historian)
- PAT knowledge management systems (a useful explanation of PAT systems is provided in Multivariate Analysis in the Pharmaceutical Industry).11
These systems operate synchronously and are used to detect and correct issues before they become problems and deviations. This is in contrast to the traditional manufacturing process, where correction is usually impossible and rework/quarantine and scrap is usually the result.
Process controls and digitized process equipment, combined with process sensors, assess the health of both the process and the product, as it exists in the process without unnecessary physical sampling. It is important to note that such systems are not about bringing the laboratory to the process, but rather about using the information to detect slight changes in materials and provide flexibility in the process operation. If only quantitative testing is the goal, this is quality by testing (QbT) and defeats the purpose of implementing technology to manage real-time control and assurance of QbD. Quantitative assessments of potency are to be considered side benefits of the QbD approach, not the major driver.
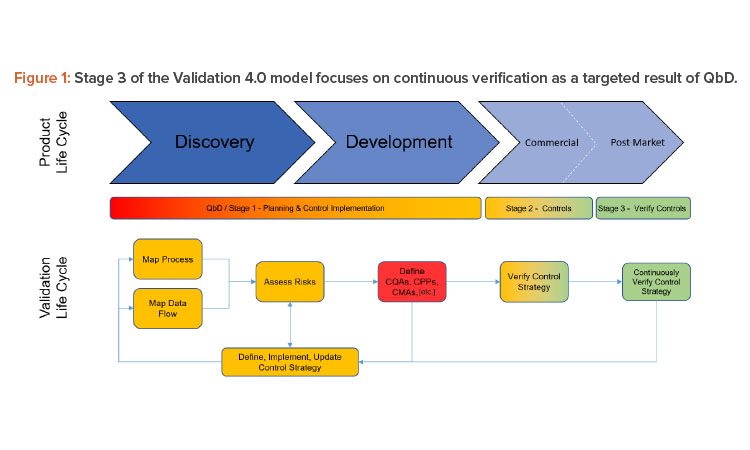
Figure 2 shows how digitized recipe systems, once integrated to process sensors and data that represents the process signature, allow a complete and continuous assessment of the entire batch, effectively making every batch a validation batch.
Case Studies from OSD Manufacture
The case studies outlined in this article are from oral solid dosage-type production; however, the principles are applicable generally and the objective of this article is to encourage similar approaches and applications across the Pharma and Biopharma industry. The following case studies are from different types of OSD manufacture and are representative for both batch and CM systems. A typical OSD process starts with raw materials, dispensing of raw materials, milling/sieving, blending, compression, coating, and packaging. The operations of granulation (wet or dry) differ with the raw material and are a particle engineering step used to provide the granule properties conducive to uniform blending. Powder blending is one of the least understood of scientific processes and is influenced by many aspects of materials, including their size, shape, density, electrostatic nature, and moisture content. Without a complete understanding of raw material characteristics, process improvement and optimization are very difficult.
Many organizations globally are starting to adopt PAT systems for a number of unit operations, particularly focusing on blend uniformity. When these companies monitor blend uniformity using technologies such as NIR spectroscopy, they are often disappointed with the results obtained. When a process is measured at the microscopic level, the flaws associated with traditional validation approaches are revealed. This may sound surprising and, in many cases, a manufacturer will argue that their product has been manufactured without variability issues for years. A quick review of batch documents, however, tends to reveal a long list of process deviations and reworks, simply because current validation strategies are short term, rather than long term and performance focused.
Case Study 1
The first case study shows why the blending stage is typically the wrong place to start with PAT for process improvement, because operations have already transformed raw materials into intermediates before blending is performed.
Some raw material suppliers provide active pharmaceutical ingredients (API) that are pre-granulated so that the secondary manufacturer can produce tablets using direct compression batch manufacturing process. During the validation stage of the process, a traditional three-batch approach was used, whereas typically the raw material supplier sends exhibition batches of their material and the manufacturer typically assigns the company’s best operators and analytical staff to perform the validation. In this scenario, the validation is already biased.
Problems start to arise soon after process validation is approved, and in one situation, the quality control (QC) laboratory allowed the material to pass based on pharmacopeia identification and purity testing. Process operators noted that the new raw material lots were free-flowing at the top of the bags, but as they started to empty the bottom, heavier screening was required. The company decided to invest in NIR spectroscopy to evaluate the process and to cope with the expected high throughputs of the new product introduction.
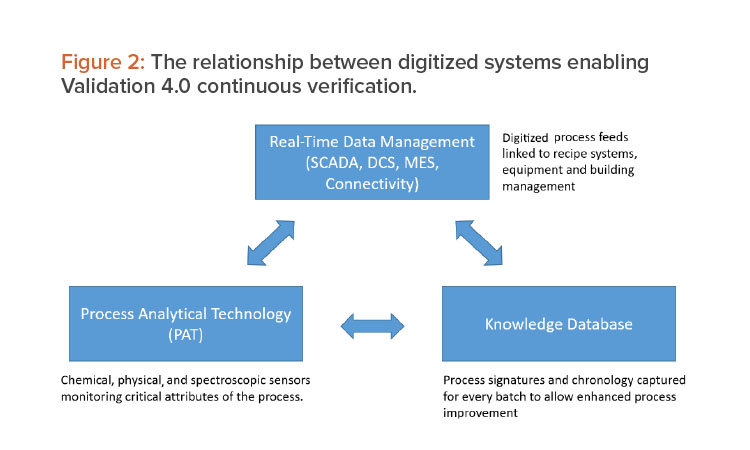
The first observation was that the NIR detected a distinct difference in the original raw material lots and the new lots, even though they passed pharmacopeia testing. Figure 3 summarizes the findings of an extensive process investigation.
Using a combined NIR spectroscopic/chemometrics approach, deviations from the expected process signature were observed. When these issues were traced back to the raw materials, it was found that the particle size distribution (PSD) of the new lots was inconsistent. NIR spectroscopy is capable of detecting median particle size changes and this inconsistency resulted in more fines, based on lots that were not blended properly that had uniformity and compression issues.
As stated previously, granulation is the process of engineering particles to tight PSDs so that they are matched to the other material, allowing uniformity in blending. This is not homogeneity because a homogeneous material is perfectly mixed and is an impossible state in reality. Better terminology is heterogeneity minimization, which is more appropriate for pharmaceutical blending operations. This example highlights the need to understand the raw material as the first stage of OSD manufacturing process validation and shows how technology can measure changes in raw materials and the characteristics of intermediates. Technologies such as NIR spectroscopy, inline particle sizers, and other sensors working in combination can be used to develop process models that assess data in real time and allow feed-forward process control.
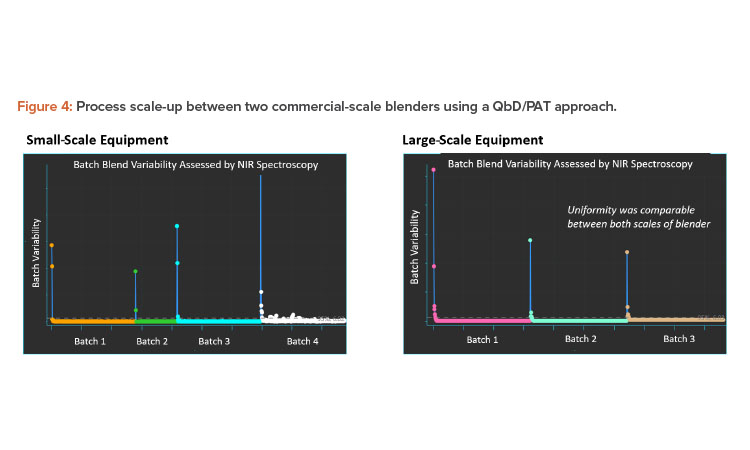
Case Study 2
A second case study focuses on the scale-up of a blending process from one commercial manufacturing batch size to a large batch size. The intermediate bulk container (IBC) geometry is similar between the two blenders, with one offset at 15 degrees and the other at 30 degrees off axis. Because the smaller scale process is considered validated, it can serve as a baseline when comparing the results to the larger blender.
Inline NIR spectroscopy was used for the evaluation and the results are summarized in Figure 4. The blend is a high-dose API formulation and can be monitored for uniformity using inline NIR spectroscopy. A spectrum is collected after each rotation of the blender and as the heterogeneity minimizes, spectra collected will begin to look consistent with each other. Statistical methods can then be applied to the data, resulting in the blend curves shown in the figure. In all cases, the endpoint was established rapidly compared to the endpoint established by traditional validation approaches.
The results in Figure 4 highlight the principles of Validation 4.0 continuous verification and why digitalization is such an important enabling tool for QbD. However, this does not negate the need for the application of suitable processing engineering mechanisms. In the preceding example, blend uniformity can be established without physically sampling the powder bed, whereas the traditional validation approach used sample thieves to extract nonrepresentative specimens from spatial locations. Not only is it impossible to representatively sample a three-dimensional lot, but the sampling errors induced by using a sampling thief contained 10 to 50 times the errors in the analytical instrumentation used to determine potency. We found that the traditional validation approach required 10 to 20 times longer blending of the product, which can induce demixing and reduction in particle size of softer materials.
This is one of the main reasons the US FDA has questioned USP <905> (Uniformity of Dosage Units)13 as a method for establishing blend and content uniformity. The shearing and electrostatic forces induced by physical sampling devices can destroy blend uniformity and provide false information on true blend endpoints. The NIR spectra provide digital information in real time of the state of powder mixing and the process can be stopped when the desired state has been reached. This is an outcome of the philosophy of PAT that must be translated to Validation 4.0, i.e., processes are ready when they are ready. Fixed-time processing is not in alignment with QbD or Validation 4.0 and technology must be used to continuously verify when the desired state has been achieved.
In one example, a manufacturer wanted to validate the NIR method against physical aliquots extracted from the blender using a spatial sampling plan. After extraction of the aliquots, the blender was restarted and the NIR spectrometer indicated that the entire uniformity of the blend was lost. Uniformity was reestablished after 60 further rotations and the original blended state was not reached. Again, digitalization and no physical sampling were able to establish the correct endpoint of the process and using PAT, a continuous verification strategy was established.
Case Study 3
In tablet compression, each tablet produced is a 100% statistically representative sample. The sample delivery of powder to the feed frame of a compression machine resembles the most accurate sampling device currently available: A spinning riffler. A rotary compression machine can be visualized as a compacting spinning riffler and therefore analysis of single tablets is a true representation of blend and content uniformity.
Because blending does not stop until compression, an opportunity exists for manufacturers to monitor uniformity in real time at the feed frame of a tablet press. In this case study, a NIR spectrometer is located on the spider wheel and is used to ensure representative sampling, and the NIR spectrometer can measure content uniformity in real time and report the data in control charts. When only random variations around a target value are observed, this can be used to establish that the blend has retained its integrity during transfer and that the content uniformity is confirmed.
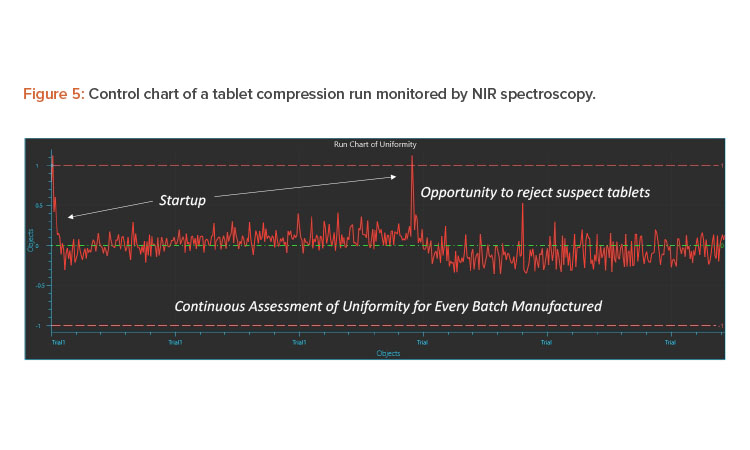
The 100% measurement of individual tablets is practically not possible; however, due to the representative nature of the sample in the feed frame, pseudo 100% verification of blend and content uniformity is established. This is an example of the true nature of the scientific, risk-based approach outlined in “Pharmaceutical CGMPs for the 21st Century—A Risk-Based Approach: Final Report”7
The European Medicines Agency has published a “Use of Near Infrared Spectroscopy (NIRS) by the Pharmaceutical Industry and the Data Requirements for New Submissions and Variations,”12 which allows PAT applications to produce trend data for quantitative analyses. In this example, the NIR results represent uniformity of blend, then by measuring at the feed frame, content uniformity can be established through consistency in control chart-ing.
The first step of monitoring at the feed frame uses digitalization to establish uniformity. The second step is a traditional approach where tablets are collected at uniform time periods over the compression run and assayed using the reference laboratory method. Because the samples obtained are representative, the first validation batch can be comparative and subsequent validation and continuous verification batches can all be monitored by inline PAT methods. Figure 5 shows the results obtained from two IBC bins run on a compression machine and assessed by inline PAT. The Y-axis can be a predicted value using a validated chemometrics model or a specific and selective wavelength that corresponds to changes in API or other important components. As is typical, during changeover of IBC blenders and tablet press startup, deviations can be observed in the control charts. Such tablets can be rejected off the line until uniformity is established and then tablets can be collected for coating or packaging.
Conclusion
The world is rapidly becoming fully digitized, and concepts like the IIoT and Industry 4.0 have led to innovations in rapid data storage and use. Validation 4.0 must leverage these concepts and fit them into established QbD and PAT initiatives currently being adopted by the pharmaceutical industry and related industries.
New PAT instrumentation with smaller footprints and higher sensitivity are constantly being developed; when used correctly, they can allow manufacturers to become innovative and reduce or eliminate the need for physical sampling and offline analysis. This allows industries to apply the principles of Validation 4.0 to control and assure the quality in real time for every batch with an applied knowledge and data-driven intelligence from historical trends. Thus, Validation 4.0 is maintaining a chronological history through data of the entire manufacturing process and when deviations are detected, these finding can be analyzed by methods, either statistical or chemometrics, to establish the root causes of the issues and develop control strategies to minimize the occurrence of such events in the future.
The key takeaway is that the principles of Validation 4.0 are proactive, not reactive. Under the old paradigm, traditional approaches were biased and based on selecting batches with the best raw material, operators, and analysts as a baseline to pass product for release. In Validation 4.0 and a truly QbD system, the use of data models, PAT, and feed-forward/feedback control establishes a process chronology and digital signature for comparison to past and future batches. Therefore, digitization and QbD allows for true validation of every batch.
Please reach out to the Validation 4.0 SIG with your questions and to share your views, thoughts, case studies, and concepts on new methods that will better assure quality in pharmaceutical and biotechnology manufacturing. The Validation 4.0 community welcomes your input.
About the Authors




