Streamlining Postapproval Submissions Using ICH Q12 & SCDM
Postapproval change management of pharmaceuticals is an essential part of life-cycle management but is associated with regulatory challenges. Incorporating concepts and tools from the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Q12 guideline, combined with structured content and data management (SCDM) and a cloud-based data exchange platform, could provide synergistic benefits that will enable efficient supply maintenance of life-saving therapies worldwide.
Following approval of an initial marketing application, postapproval changes are needed to ensure adequate supply, mitigate supply risk, expand patient market access, optimize manufacturing processes, improve analytical methods, and comply with new regulatory expectations. Regulatory submission and evaluation of chemistry, manufacturing, and controls (CMC) data may be required for changes that have a higher risk to impact product quality. Such changes can include increasing batch sizes, adding manufacturing facilities, creating new product presentations, and updating analytical methods. Prior to implementation of a change, manufacturers conduct risk assessments and generate data to confirm there would be no adverse impact to product quality through shelf-life as a result of the change.1, 2
Health authorities have specific regulations and guidelines that govern reporting requirements necessary to implement postapproval variations; however, regulatory submissions for a given change can vary based on differing regional requirements as specified by the health authority conducting the review. For changes requiring approval before implementation, once the necessary information has been submitted across regions, each health authority must review the data package and documentation based on local requirements.
The timing and data requirements vary depending on the reporting category, submission, and legal obligations in each individual jurisdiction. For products with a large global footprint, the time required from submission to global approval and implementation of a single change can take years while the change obtains approval across all the relevant regulatory agencies. The resulting complexity requires manufacturers to tightly control and manage supply chain activities for commercial products globally to maintain compliance. 1, 2 Depending on the nature of the change, manufacturers may be required to maintain both the pre-and post-change equipment, processes, or methods until all approvals are received.
In an attempt to address these challenges, in 2019 the ICH endorsed ICH Q12, Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management (and Annexes). This guideline was developed and designed to provide a flexible framework to facilitate postapproval CMC change management, and outlines a risk-based, structured, and harmonized approach.3, 4
The current lack of global harmonization for CMC content across regions requires the burdensome task of creating multiple submissions to fulfill varying regulatory requirements across different markets. Typically, CMC information needed to support a postapproval submission is accessed and transcribed manually from raw data systems (e.g., laboratory notebooks, batch records, and technical reports) to submission documents (e.g., Common Technical Document [CTD] sections). This information package is then submitted in electronic PDF format that health authorities must manually “unpack” or deconstruct in order to perform their assessments. These efforts consume substantial time and resources for both sponsors and health authorities, which may result in redundancies and the need for multiple data integrity checks. The use of SCDM can potentially help alleviate the burden of submission authoring by shifting the focus from document management to data management, including how information is stored, analyzed, authored, and reviewed.2, 5
This article will examine the current global regulatory submission workflow for postapproval CMC changes and will propose principles to streamline regulatory submission authoring through the utilization of ICH Q12 concepts, in combination with SCDM. It will also discuss several challenges associated with full implementation of the ICH Q12 regulatory tools, including established conditions (ECs), postapproval change management protocols (PACMPs), and product lifecycle management (PLCM) documents. Other challenges addressed in this article include the varying interpretation and lack of standardization that must still be overcome to achieve true global harmonization. This review also considers the next steps toward achieving simultaneous global regulatory submissions and utilizing cloud-based technology to facilitate data exchange between sponsor and regulator.6
ICH Q12 Global Implementation: Status and Challenges
The ICH Q12 guideline provides a framework to facilitate post-approval CMC changes more predictably and efficiently through a unified risk-based approach that will benefit patients, the industry, and regulatory authorities.3, 4 Full implementation of ICH Q12 tools across all jurisdictions may be delayed until the necessary legislative changes are made.7, 8, 10 ICH Q12 introduces the concept of established conditions (ECs), which provides a clear understanding between the manufacturer and health authorities regarding critical manufacturing, quality, and analytical elements that require regulatory actions.
In May 2021, the US Food and Drug Administration (FDA) published a draft industry guidance, ICH Q12: Implementation Considerations for FDA-Regulated Products. This guidance complements ICH Q12 guidelines by clarifying how the ICH Q12 tools and enablers can be implemented within the US regulatory framework, and reflects critical lessons learned from the 2019 FDA Pilot Program on Established Conditions. 9, 10
The European Medicines Agency (EMA) issued its implementation guidance in March 2020.10 It emphasizes that one must always default to the requirements laid down in the current European Union (EU) variations of regulation and associated guidelines.6 Currently, the EU legal framework does not recognize the product life-cycle management (PLCM) document, which contains ECs and proposed reporting categories but does recognize PACMPs. The European Commission, together with the EMA and the National Competent Authorities, will continue to work on fully implementing the ICH Q12 guideline within the existing EU legal framework7, 11, 12
In Japan, the Pharmaceuticals and Medical Devices Agency (PMDA) introduced minor change notifications to the regulatory framework. However, there was no harmonization of post-approval change reporting categories across the ICH regions at the time. The application form, unique to Japan, can be the basis for using ECs to determine filing strategies for postapproval changes. Due to this unique approach, standardized global implementation of ICH Q12 has been challenging.13 In an effort to achieve global standardization, the PMDA released a notification in March 2021 that confirms the incorporation of PACMPs into the regulatory framework, and full implementation was expected soon at the time of this writing (late 2021).14
At ICH Day during DIA China 2021, industry and regulatory experts agreed that real experiences from well-established regulatory systems would help China implement ICH Q12 from policy and technical perspectives. At the regulatory level, the National Medical Products Administration (NMPA) Drug Administration Law (DAL), Drug Registration Regulation (DRR), and Provisions for Post-Approval Changes have provided support for the transformation and implementation of ICH Q12 in China. The FDA has also offered both early dialogue and training to share knowledge and experience with the industry, providing an excellent example for other regulators to follow. Based on these examples, the industry in China will look to NMPA to help guide and create similar tools for effective implementation.15
In July 2021, Health Canada (HC) released an updated draft guidance for its Post-Notice of Compliance (NOC) Changes and solicited public comments. The draft presents new guidance on what information should be submitted to HC to set ECs and proposed reporting categories, and requirements for PACMPs.16 As part of HC’s implementation of ICH’s Q12 guideline, HC announced a pilot program that is specifically seeking applications and supplemental applications for biologics and pharmaceuticals that will use ECs and PACMPs.17 HC is expected to have full implementation of ICH Q12 by late 2022.18
It is anticipated that even after full implementation of ICH Q12 in each region, varied health authority interpretations of the regulatory tools could lead to divergence in the approved ECs across jurisdictions, as different ECs and proposed reporting categories might be approved. Disagreement on ECs and their reporting categories could result in extended negotiations, potentially delaying review times.19 ICH has developed additional training materials for ICH Q12 to benefit both the industry and regulators. In the future, more guidance and experiences gained through participation in pilot programs, such as the FDA EC pilot, may be necessary to clarify some of the ambiguity in identifying ECs.17 Recent engagements between industry leads and health authorities have highlighted the value of using a standardized approach for the regulatory tools in ICH Q12.10 However, the long-term expectation is that the use of ICH Q12 and ECs will decrease the need for postapproval filings and regulatory agency interactions.20, 21
Implications for Existing Processes
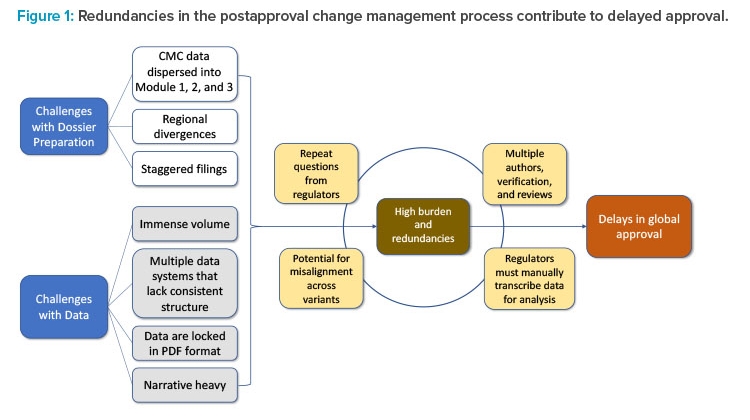
Delays in global approvals experienced due to lengthy CMC postapproval regulatory processes can be traced to two factors: challenges with dossier preparation and challenges with data. These challenges exist because of outdated regulatory systems and the inability to leverage state-of-the-art technology for exchange of information such as data and narratives. Collectively, challenges such as the spread of data across multiple document sections, regional differences, staggered filing timelines across markets, and the immense volume of data locked in PDF format contribute to the high burden and redundancies in postapproval regulatory submissions. The accumulation of these redundancies contributes to delays in implementation of necessary post-approval CMC changes and discourages innovation.
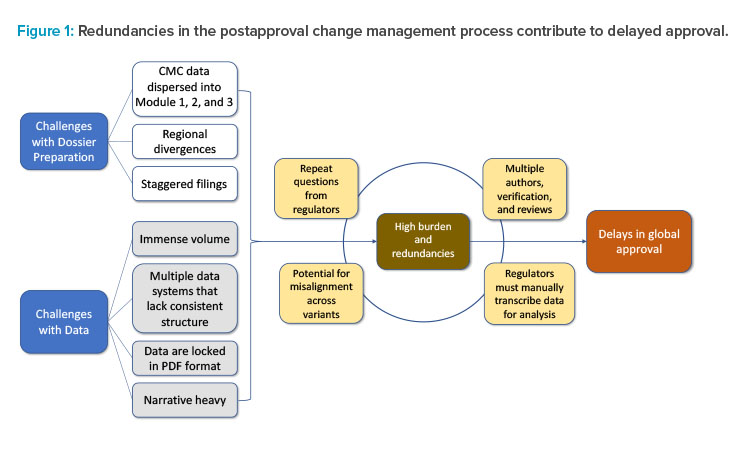
The current CMC authoring process for a regulatory dossier requires writing, reviewing, verifying, and approving multiple sections and subsections within Modules 1, 2, and 3 (Figures 1 and 2). This information is often spread across separate documents within a section of the module and must be manually entered, often in multiple places, before reverifying data and reapproving. This arduous compilation process limits the ability to compare data collected for different products and submissions. In addition, if the information provided by the sponsor requires updates or is sent to different regulators, it must be manually updated and reverified in each document in which it appears, as information is not conveniently linked across documents to enable real-time editing (Figure 1). As a result, multiple regional variants of each document must be maintained and robust tracking efforts are required to ensure that each health authority receives accurate and updated information.2
It is similarly challenging for health authorities to provide consistent product feedback, as accessing sources, documenting prior decision-making, and review documents may also require manual work. Thus, the current submission and review process must transition from managing documents to managing data to address these challenges, remove unnecessary labor and repetition, and create more time for scientific rationale and risk-benefit analysis.5
Numerous steps are involved in the authoring process, with the integration of ECs into the process. The postapproval change authoring process involves many stages of writing, review, and verification for large amounts of data. SCDM implementation can potentially assist with streamlining the development and verification of data necessary for these regulatory submissions.
SCDM provides the framework for authoring human- and machine-readable content by enabling an enhanced review, organization, and standardization of electronic narrative and data, allowing uniformity of approved information across documents.5 Data automation with SCDM can potentially reduce errors and increase the accuracy of data across submissions. SCDM systems could improve the efficiency and quality of regulatory documents by establishing a standardized authoring process, mitigating risks, and decreasing time to implementation. Once the SCDM system is capable of this level of real-time data automation, the flow of existing data to finalized documents that are ready for health authority review can be established. Ideally, data and information from multiple regions could be submitted simultaneously, available for all regulatory bodies to access and review as required, thereby allowing regulators to communicate with each other and see previous questions and decisions of other regulatory bodies.
Substantial resources are required to develop and implement these advanced SCDM solutions, which has been a barrier to becoming the industry standard. SCDM could be especially useful for CMC data as it allows for creating, capturing, and reusing component information as product development progresses. Coupling the integration of ECs with the design and implementation of SCDM may increase postapproval process efficiency and enable some authoring automation. It would also ensure the ECs—which can be captured in multiple different CTD sections including Module 3 sections, Module 2 sections, and the PLCM document—are aligned. 2, 3
SCDM systems could improve the efficiency and quality of regulatory documents by establishing a standardized authoring process, mitigating risks, and decreasing time to implementation.
Case Studies
Case Study 1: Potency Assay Change
Assessing potency of a drug product is a critical quality attribute of biological therapeutics.22 Frequently, potency assays for biologics are in vitro cell-based and have complicated mechanisms of action intended to parallel that expected in vivo. Though powerful tools, cell-based bioassays can be challenging and may require remediation to increase robustness and operational need (e.g., risk mitigation of critical reagents/instruments). It is a complex task to implement a modification to a potency assay across different regions as prior regulatory approval would typically be required.
In this case study, a company is interested in optimizing a potency assay for a therapeutic monoclonal antibody approved in 50 countries to decrease the rate of invalid test results. As part of the change management process, personnel must manually find, view, and interpret the older data sets to establish a frame of reference for completing method validation exercises and compare the initial and optimized assays. Following data collection, the authoring and submission process begins as described in Figure 2, which could take a minimum of three months to complete. Testing with both potency assay methods, pre- and post-change, is necessary to gain approval from health authorities, resulting in two sets of data to be collected and submitted to each health authority.
As the product has an extensive global footprint, full approval for the change across all jurisdictions could take as long as five to six years. During this time, duplicate testing with both potency assay methods would be necessary, resulting in a significant cost burden to the sponsor. Incorporating ECs into this case study, when agreed upon with the regulator, can potentially lower the reporting category of the change, thereby reducing the timeline for approval (Figure 3). The use of a PACMP may be another option where similar timelines may be achieved. As a result, in this example, the applicant might implement the new potency assay method after 30 days for the US (CBE-30) and report the change to global health authorities afterwards in a more structured timeframe. Providing data in a usable SCDM format through a cloud-based ecosystem would have the potential to enhance data analysis, further improve global filing and review efficiencies, and shorten the overall time to global approval.
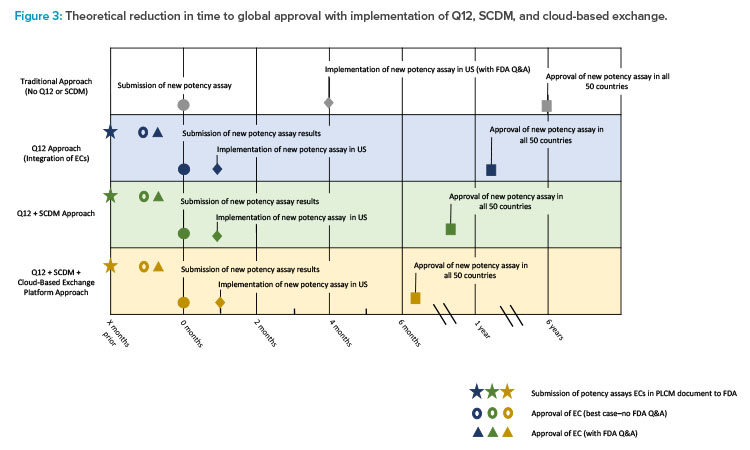
The first row of the timeline (white) displays the approach taken without incorporating ECs or SCDM. The second row (blue) demonstrates how the use of ECs can lower the reporting category. The third row (green) demonstrates the shortened time to global approval that can be achieved by incorporating both ECs and SCDM.
Additional efficiencies and further reduction in time to global approval are possible by leveraging a cloud-based filing and review platform (fourth row, yellow).10 For example, two large data sets could be automatically imported into regulatory documents without manually transcribing and locking data in PDF format. The use of ECs can streamline the postapproval process by potentially reducing the reporting category of the change, while SCDM may increase the efficiency and automation of document generation for submission to multiple countries. The use of both ECs and SCDM could thus shorten the timeline for global change approval and implementation.
Case Study 2: Alternate Sterile Filter to Address Shortage
Filter shortages across the industry due to the COVID-19 pandemic are driving the need to rapidly qualify new filters from alternate vendors. All filters used in the manufacture of a product must be characterized by the pharmaceutical quality system (PQS). The characterization data need to demonstrate that the filters are equivalent, with no impact to bacterial retention, product binding, or the extractables/leachable risk profile.
According to existing guidance, prior approval is required to implement an alternate filter in most markets. Due to varying regulations and long approval timelines, global implementation of an alternate filter may lead to delays in implementation, and potentially lead to drug shortages.
Due to the nature of the change and potential impact to product quality, and per a sponsor’s internal PQS, process validation data are required to support a filter change. In this case study, the sponsor submits a PACMP to a health authority. The PACMP contains small-scale characterization data for the alternate filter, with a commitment to provide at-scale data in subsequent annual reports. The filter validation acceptance criteria are ECs in the previously approved PLCM.
In this scenario, if the sponsor has an approved global PLCM document describing the ECs with a reduced reporting category, the time required to implement the alternate filter can be reduced. Identical submissions are created with the same viral filter load in-process controls (IPCs) and submitted to all regions with a reduced filing category and overall implementation timeline. However, information requests from various health authorities invariably lead to additional country-specific variants containing different acceptable ranges for viral filter load IPC, thus creating multiple variants of Module 3 sections and PLCM documents.
The use of SCDM in this case study could allow data from previous filter characterization studies, including cleaning validation, viral clearance, and extractables/leachables to be better tracked to improve efficiency of the authoring process. Filing preparation would be simplified, as data would not have to be manually transcribed and verified, saving on submission authoring time. When information requests from various health authorities are received, filing automation would allow efficient updates to Module 2, Module 3, and the PLCM document. Thus, use of ECs together with incorporation of SCDM concepts would shorten the timeline for change implementation, thus de-risking potential impact on patient supply.
Case Study 3: Trypsin Reagent Replacement for Peptide Map Method
Routine method revisions enhance method performance and mitigate critical reagent supply. In this theoretical case study, the sponsor must replace the trypsin reagent used in the peptide map method due to a raw material shortage. The sponsor is required to evaluate an alternate manufacturer for trypsin and to characterize the new trypsin (from the same source),23 which is then qualified, confirmed equivalent to the previous trypsin, and deemed acceptable for use. Execution of the peptide map method does not change in response to the new trypsin that is filed globally; however, some regions may require prior approval to implement this change. The sponsor incurs unnecessary expenditures by making this change because they must run peptide map testing twice, using both the old and the new trypsin, until global implementation of the change can be achieved. The sponsor also runs the risk of running out of stock of the currently approved trypsin, potentially leading to a supply risk in countries where the change has not been approved.
Using ICH Q12 principles, the peptide map method would be filed with an accompanying PLCM document in which any low-risk change to raw materials in a method is defined as an EC with a “notification low” reporting category, meaning the change would not require approval or notification prior to implementation. The required qualification data justifies the lower reporting category. With prior regulator agreement, this method reagent change can then be implemented as a notification low/annual reportable change with reduced reporting categories worldwide. The sponsor can implement the new trypsin immediately in drug substance/drug product (DS/DP) release testing with one peptide map method, eliminating costs associated with redundant testing. The old method can be replaced immediately, and duplicate testing would not be required.
However, the sponsor is still required to author the submission and submit the data for the peptide map raw material change to each region. As described earlier, creation of individual submissions for each region, including country-specific variants, is a resource-intensive process. The overall time required to author and submit the trypsin reagent change could be reduced by leveraging both ICH Q12 tools as well as SCDM and authoring automation.
Use of ICH Q12 methodology would lower the reporting requirements, allowing for quicker implementation of the change. SCDM would decrease resources and time needed to manually transcribe and verify data in a CTD section. SCDM would also allow for rapid exchange of additional data if health authority reviewers needed it to support review.
Future Considerations
During the COVID-19 pandemic, it has become apparent that the pharmaceutical industry can rapidly adapt in response to public health emergencies. The fragile nature of the pharmaceutical supply chain became apparent, demonstrating the need for manufacturing flexibility for existing products. The rapid transition between clinical studies to emergency use authorization, and then to the world’s first fully approved vaccine in under two years, highlights the potential of biopharmaceutical companies and health authorities. On the basis of medical need, science-driven risk, and the need for manufacturing flexibility and efficiency, similar acceleration of regulatory approvals could potentially be achieved for any life-saving medication. The technology now exists for information and data exchange within the postapproval space to match the speed of product drug development and innovation.
To meet the challenges in the existing CMC regulatory landscape, standardization and harmonization of ICH Q12 regulatory tools across different regions (including non-ICH regions) would be needed. In addition, significant regulatory reform, and modernization, including digitalization and digitization, would be necessary to achieve a single standardized global submission. At the FDA, the Technology Modernization Action Plan (TMAP) and the Data Modernization Action Plan (DMAP) aim to modernize its digital infrastructure.24 The FDA has recently announced the reorganization of the agency’s information technology (IT), data management, and cybersecurity functions into the new Office of Digital Transformation (ODT). The ODT allows more effective data management to streamline operations by reducing duplicative processes and implementing technological efficiencies.25
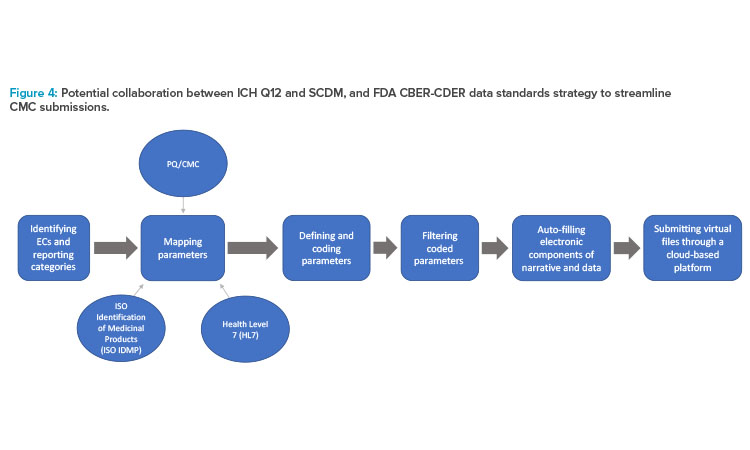
The flowchart in Figure 4 shows the overlap and proposed workflow after both the sponsor and the health authority implement ECs; SCDM; identified and mapped data parameters based upon FDA PQ/CMC (pharmaceutical quality/chemistry, manufacturing, and control) information; and universal standards, such as International Organization for Standardization (ISO) Identification of Medicinal Products (IDMP) and Health Level 7 (HL7) standards.
With the implementation of ECs and SCDM, sponsors could standardize and simplify their internal documentation while maintaining alignment with the externally standardized regulatory requirements. In that case, data and information can be further coded and standardized into an online, cloud-based data exchange platform that can facilitate automatic updates.26,27,28
The FDA’s Center for Biologics Evaluation and Research (CBER)/Center for Drug Evaluation and Research (CDER) Data Standards Plan is an ongoing project that aims to map PQ/CMC data elements, standardizing application content to facilitate efficient risk-based reviews by linking data and common categories and elements across various application types. A secondary goal of this project is to provide recommendations for standardization of the categories and elements necessary for application review. Where corresponding data elements exist, the identified PQ/CMC data parameters overlap and, in some instances, directly align with the substance and product identifiers described by the ISO IDMP standards (Figure 4). After the identification and standardized mapping of these data parameters, they are submitted to the ICH CTD.24,29 The longer term goals of this project are to increasingly replace dossiers with structured content and data supporting dossier variations with more efficient online database updates.28
The need for a more templated and structured approach to the sections within Module 3 is clear. Revision of ICH M4Q has commenced with endorsement of the concept paper in November 2021, to be followed by an ICH topic proposal on structured product quality submissions.23 Both ECs and SCDM facilitate standardized language across regions, and this language can align with mapped data parameters using PQ/CMC, ISO IDMP, and HL7 standards and Fast Healthcare Interoperability Resources (FHIR) artifacts. The collaboration between these tools can allow sponsors to store structured information to potentially be used as a single global submission that is consistent, reproducible, and easily accessible by health authorities across different regions.24, 29, 30
To achieve the goal of a single global submission, up-to-date information must be exchanged seamlessly between sponsors and regulators. Accumulus Synergy, a nonprofit sponsored by leading biopharmaceutical companies, is developing a cloud-based platform intended to facilitate real-time data exchange and review in a worldwide setting. The platform under development proposes locked and shared spaces in the cloud, allowing both sponsors and regulators to work and communicate with each other across portals and protected by firewalls. Sponsors could use SCDM systems to automate the compilation of data and electronic narrative and push this information to the Accumulus cloud to facilitate efficient data exchange, collaboration, and parallel reviews, thus improving the submission and review process.
In addition to ease of access and exchange of information, reviewing regulatory submissions in parallel can reduce the total time to approval for a drug product in multiple regions. Providing a platform by which data can be exchanged in usable format enables more efficient filing processing and improved assessment capabilities. Once the principles of data exchange automation are established, they can be extended to clinical, preclinical, and summary data.6
Conclusion
There is a perception that once a product is commercialized, or a marketing application is approved, that the work associated with product filings is complete. On the contrary, the work is essentially only beginning from a CMC perspective because the product is optimized multiple times and in many areas over the lifetime of its commercial viability. Several challenges burden the current global regulatory submission workflow for postapproval CMC changes. There is a need for efficiency and consistency within the postapproval regulatory space to reduce the high costs, complicated surveillance of updated regulatory requirements, and management of the large volumes of data that accompany CMC changes.
The use of regulatory tools, namely ECs as described in the ICH Q12 guideline, provides a unified and flexible approach in reporting postapproval changes to health authorities, and an avenue for more efficient change implementation. Combining ECs with an SCDM system may enable automated authoring of human- and machine-readable content supporting an enhanced review, structured organization, and standardization of documents submitted to health authorities in line with the PCLM. With the varying interpretation of the ICH Q12 guideline by sponsors and health authorities, efforts must be made to enhance standardization and harmonization. As a complement to the initiatives taken by the FDA, SCDM could be an efficient solution to address challenges in the current CMC regulatory submission and review process. Combining the benefits of ICH Q12 and SCDM with a cloud-based filing and review ecosystem could propel the pharmaceutical industry digitally forward to deliver therapies more effectively and efficiently to patients around the world in a timelier manner. These concepts and platforms can subsequently be leveraged to expand beyond CMC data and attain a single global regulatory submission for new drug applications including non-clinical, clinical, and safety modules.
Reader note: This article was initially submitted to Pharmaceutical Engineering® during December 2021/January 2022.
About the Authors


Acknowledgements
The authors acknowledge Rita Algorri and Andrew Lennard for providing comments on the manuscript.


