Continuous Viral Vector Manufacturing

A growing segment of the advanced therapy medicinal product (ATMP) landscape, which includes gene therapies and cell-based treatments, relies heavily on viral vectors for efficient gene delivery. The increasing demand for these therapies requires a robust, scalable, and cost-effective manufacturing solution.
Traditional fed-batch viral vector production poses challenges such as variability, long processing times, and limited scalability, making it difficult to meet commercial and clinical demands. Continuous biomanufacturing (CBM) offers a potentially transformative approach by enabling consistent product quality, reducing processing time, and improving overall efficiency.
Implementing CBM for viral vector production requires careful integration of key unit operations, including viral inactivation, purification, and formulation, while ensuring regulatory compliance and process robustness. By leveraging continuous processing strategies, manufacturers can enhance the scalability, reproducibility, and accessibility of ATMPs, ultimately accelerating their path to patients.
Background
There is a rising demand for high-quality viral vectors due to their promising therapeutic applications. In the field of vaccination, novel vaccines are being developed to address new pathogen outbreaks, and rapid and more efficient processes are required to respond as fast as possible to this demand. Viral vector–driven gene therapies have been proven efficient and safe in numerous clinical trials, and some viral vectors are already approved for commercialization. However, viral vector production is still a challenge on the pathway to clinics. Batch and fed-batch culture modes are preferred in the biologicals manufacturing industry.
Continuous manufacturing has been proven to improve viral titers and reduce the bioprocessing time and costs. In the context of viral vector production, CBM offers a number of promising advantages in terms of efficiency, scalability, and cost-effectiveness compared to traditional batch processes. Figure 1 demonstrates how continuous manufacturing can be applied to viral vector production, its potential improvements, and the challenges associated with implementation and operation..1
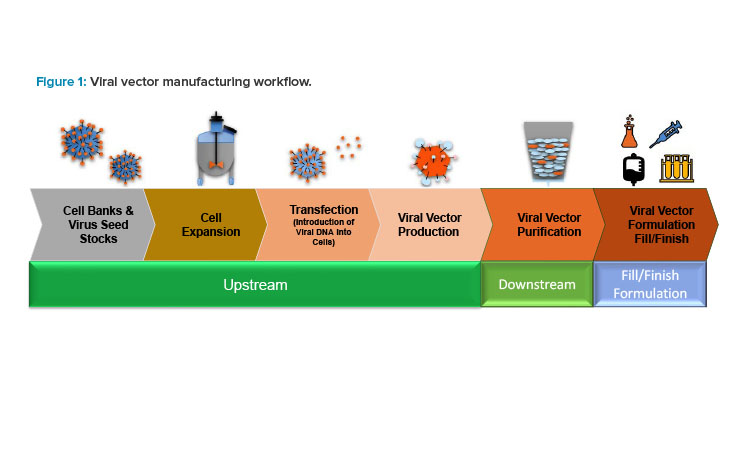
The interest in viral vector research and development has steadily increased over the years, but production processes have been given only minor attention. The development of efficient, scalable, robust, and reproducible production processes is required to meet the increasing needs of viral vectors for gene therapy programs at a reasonable cost. The most used production systems are still batch or fed batch, most often based on adherent cells (i.e., requires attachment to a substrate or surface for growth and proliferation).
There are key criteria that shape the different viral vector production systems (see Figure 2).2
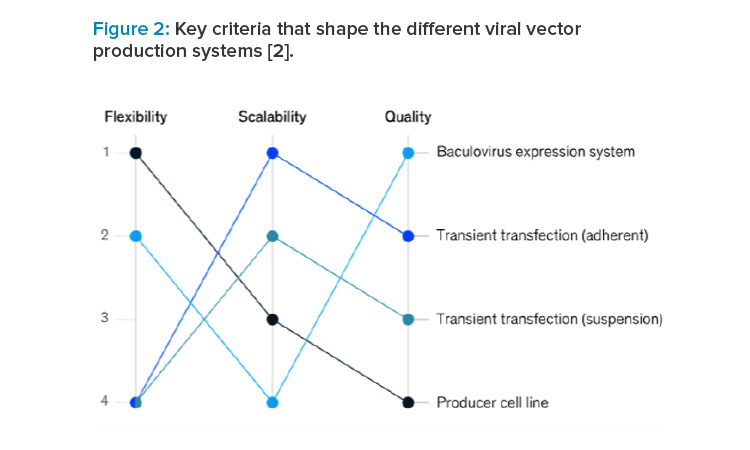
Batch production of viral vectors typically suffers from low titers.3 This is because viral vector manufacturing includes the following challenges.
Scalability
Producing large amounts of high-quality vectors for commercial applications for these products typically targets smaller patient populations. Batch sizes are much smaller than for most monoclonal antibodies (mAbs) therapeutics and often require smaller manufacturing systems.
Consistency and Stability
It is a challenge to transform the upstream process of viral vector production into a fully integrated continuous manufacturing platform that ensures the vectors remain consistent and stable over time. The transfection mix generated from plasmid DNA and transfection reagents has a limited stability. Transfection reagents that yield more stable transfection mixes are required to perform transient transfection in a truly continuous operation.
Choosing the Right Production System
Choosing a technology platform is a crucial decision developers must make early in the process. Both adherent and suspension technologies come with their own set of advantages and challenges. Posing the right questions can help guide the evaluation and selection process:
- What is the target patient population for the disease indication? Could scalability pose a challenge during commercialization?
- Which cell lines and serotypes are planned for use?
- At what stage is process development? Is the project budget sufficient to accommodate process optimization or a potential platform change?
- What is the manufacturing strategy for both clinical and commercial stages?
- Does the regulatory filing strategy prioritize speed to market or achieving best-in-class status? Is a parallel strategy feasible, and should it be considered?
Developing Standardized Methods
Choosing the right production system, optimizing downstream processing, and developing standardized chemistry, manufacturing, and controls methods and quality assays continue to be challenges facing continuous viral vector manufacturing.
Manufacturing Baseline
Why would there be an interest (or need) in moving into a continuous manufacturing platform for viral vector production, especially given that it would likely pose a new set of challenges and risks for many manufacturing companies? The following sections help answer that question by looking at the key advantages.
Scalability and Improved Throughput
CBM offers a seamless, scalable process by maintaining continuous cell culture growth and viral production. This scalability allows for a more efficient transition from preclinical to clinical and commercial production without requiring the major overhauls seen in batch processes. Additionally, the ability to increase the flow rate (by avoiding the stop-clean-restart) by increasing the runtime of materials through the system increases the overall throughput, which is essential for meeting the increasing demand for gene therapies.
Scaling up viral vector production in gene therapy is traditionally constrained by the limitations of batch-based manufacturing, where each cycle requires time for preparation, execution, and cleaning between batches. This can result in inefficiencies when scaling to the larger volumes required for clinical trials or commercial use driven by market needs.
Speed to Market
In clinical settings, where time-sensitive treatments are often required, the ability to produce viral vectors more quickly can significantly reduce lead times for therapy administration during clinical trials. This ability to scale up production rapidly without compromising quality is crucial in both the clinical trial and commercialization phases. Continuous manufacturing enhances speed by eliminating production delays between batches. Because the process operates in an ongoing, uninterrupted flow, it enables faster production and immediate quality control.
To implement continuous manufacturing in viral vector production, some key process elements should be the focus of optimization efforts to ensure success. These key stages in the manufacturing process that benefit from continuous operations will be discussed.
Upstream Cell Culture
The upstream process for viral vector production begins with the cultivation of mammalian cells—such as human embryonic kidney (HEK) 293 cells—that are transfected with plasmids encoding the necessary genes for vector production. In continuous manufacturing, cell culture can be maintained in perfusion bioreactors, which continuously remove waste products and add fresh nutrients, allowing the cells to remain in a productive state longer than in traditional batch-based processes.
The perfusion system ensures that the cell culture environment is optimal, reducing the need for frequent harvests and enabling higher cell densities, leading to increased viral vector yields. Key critical process parameters (CPPs) such as oxygenation, pH, and temperature are tightly regulated to ensure optimal cell growth and viral production.
Downstream Purification
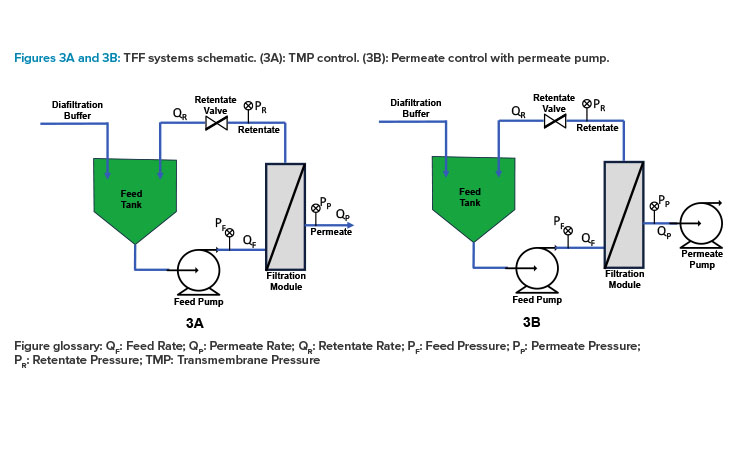
After viral vectors are produced in cell culture, they must be purified to remove cellular debris, unsecreted spent media components, and other impurities. Continuous downstream processing involves the use of filtration and separation technologies such as tangential flow filtration (TFF) and chromatography, which have been adapted to work in a continuous flow mode.
In TFF, the viral vectors are retained and are passed through the filtration machine to remove impurities. These systems can be scaled up with minimal changes, and their ability to continuously purify the viral product ensures consistency and efficiency throughout the production process. Permeate-control TFF systems are commonly used in microfiltration (MF) applications where highly permeable membranes can result in excessively high flux, compromising performance and stability. For tighter ultrafiltration (UF) applications, like those requiring 30 kilodalton (kDa) membranes for antibody retention, transmembrane pressure (TMP)-control systems serve as an alternative. On the other hand, membrane cutoffs used in viral vector manufacturing, such as for adeno-associated virus (AAV), typically range from 100 to 300 kDa. These membranes offer a balance between the tighter UF membranes and the more open MF membranes, as illustrated in Figures 3A and 3B.
Manufacturing Optimization
Drivers of the biomanufacturing industry are increasingly interested in the implementation of CBM because of optimization. This includes creating more with less via process intensification, reducing cost of goods for new therapeutics, providing flexibility for a changing market dynamic, and ensuring the ability to meet the growing demands of the industry for products. One attribute of continuous processes is the higher cell densities that can be achieved due to the continuous feeding of media, which often translates into higher production volume. Although the transfection that generates viral vectors differs from the traditional batch-driven protein expression, the cells must have a higher cell density to achieve the desired robust results.
There have been a number of detailed studies performed that align with the platform unit operations for viral vector manufacturing related to the production of mAb platforms.4 Quite often, flexibility has led organizations to see that continuous manufacturing is a way to better manage complex portfolios such as viral vectors. Viral vector manufacturing is the process of creating carriers to transport therapeutic genes, which are used in gene therapy and other fields. The process involves several steps, including the following.
Vector Design
Vectors are the vehicles used to deliver genetic material into cells. Some considerations include selecting the vector type for specific applications, designing genetic materials for specific vectors, selecting promoters for specific genes, incorporating safety features, and creating an efficient production process that includes purification and quality control testing.
Cell Culture
This step involves growing cells in a cell culture system and co-infecting them with helper plasmids. An example of cell culture in the context of co-infecting cells with helper plasmids would be the production of viral vectors for gene therapy. In this process, mammalian cells—such as HEK293 cells—are cultured in a controlled environment. These cells are then coinfected with both the viral vector plasmid (which carries the gene of interest) and a helper plasmid (which provides the necessary functions, such as proteins or enzymes) to aid in the production of the viral particles. The helper plasmid supports the replication and packaging of the viral genome, enabling the generation of functional viral vectors that can be used for gene delivery in therapeutic applications.
Purification
Purification is the process of removing unwanted materials from vectors. An example of purification in the context of viral vector production would be the use of a chromatography technique, such as affinity chromatography, to isolate the viral vectors from the culture media. After the cells have been coinfected and the viral vectors have been produced, the culture supernatant contains a mixture of viral particles, host cell debris, and other impurities. To purify the viral vectors, the supernatant is passed through a chromatography column with a resin that selectively binds to the viral particles. The bound viral vectors are then eluted, separating them from unwanted materials like proteins, DNA, and other contaminants. This process helps ensure that the final product is a pure viral vector, ready for use in applications like gene therapy.
Quality Control Testing
Quality control testing ensures the vectors are safe and pure. The implementation of CBM for large molecule viral vector drug substances requires establishing reliable analytical methods to ensure consistent product quality, safety, and efficacy. These tests should be optimized for continuous manufacturing and may require more frequent sampling or inline monitoring as the process runs continuously. The goal is to ensure robust quality control, minimal variation in product attributes, and fast detection of any process deviations.
Some of the baseline analytical tests that are commonly employed to support CBM include:
Integrity and potency assays: Functional assays, such as infectivity assays or transgene expression assays, ensure that the viral vector is intact and capable of delivering the therapeutic gene as intended. These assays often assess how well the viral vector can transduce target cells and express the desired gene product.
Purity analysis: This is an analysis of contaminants such as host cell proteins, DNA, and other impurities typically done by techniques like SDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis), western blot, high-performance liquid chromatography (HPLC), or capillary electrophoresis. These ensure the viral vector is purified and free from unwanted biological material.
Endotoxin testing: This testing includes Limulus amebocyte lysate assay or recombinant Factor C (rFC) assay to determine endotoxin levels. Endotoxins can trigger immune responses, so it’s important to monitor and control them during production.
Viral vector stability testing: Stability studies (e.g., accelerated stability testing) can assess how the viral vector maintains its integrity, potency, and identity under various storage conditions. This helps in understanding the shelf life of the drug substance produced via CBM.
Determination of titers: This is the quantification of viral particles—e.g., using quantitative polymerase chain reaction (qPCR), enzyme-linked immunosorbent assay (ELISA), or infectivity assays—to assess viral vector concentration. This is critical for monitoring and controlling the amount of viral vector being produced continuously.
Immunogenicity testing: This testing assesses the potential for the viral vector to induce an immune response. This could involve cell-based assays, such as T cell activation or neutralizing antibody titers, to evaluate the potential immunogenicity of the viral vector.
Selecting the Right Viral Vector
Adenoviruses (AVs), AAVs, and lentiviruses (LVs) are three types of commonly used viral vectors for gene delivery. These recombinant viral systems have the ability to infect a broad range of hosts, including dividing and nondividing cells, without integrating with the host genome. However, there are several key distinctions between them, including packaging capacity, level, onset and duration of gene expression, and immune response.
Viral vector selection plays a crucial role in the success of continuous manufacturing processes, particularly in the production of gene therapies, vaccines, and other biologics. The choice of viral vector impacts not only the efficiency and scalability of the manufacturing process, but also the quality, safety, and functionality of the final product. The following section gives a detailed description of how viral vector selection influences continuous manufacturing.
Scalability and Yield
Continuous manufacturing relies on the ability to produce large quantities of a product in a streamlined and uninterrupted process. The viral vector used must be compatible with scalable production systems to achieve high yield without compromising quality. For instance, certain viral vectors like AAV or LV are typically used in gene therapy applications. However, their yield can be challenging to scale due to their complex genome packaging requirements or their need for specific host cell lines.
Choosing a viral vector with well-understood behavior in continuous manufacturing systems can reduce bottlenecks and increase overall productivity. For example, vectors that require fewer components for packaging (like LV) may simplify scaling, whereas more complex systems (like AAV) might need more specialized conditions or higher-efficiency production platforms.
Cell Line Compatibility
Viral vectors are typically produced in mammalian cell cultures, and the compatibility between the viral vector and the chosen cell line is vital. Continuous manufacturing systems require cell lines that can grow in large-scale bioreactors, where conditions need to be tightly controlled for high productivity and long-term cell viability. Some viral vectors may require specific helper plasmids or co-infection with additional factors to facilitate the production of viable viral particles. The more optimized the cell line and viral vector combination, the better the continuous production process will perform in terms of consistency and output.
For example, the HEK293 cell line is commonly used for LV production due to its high transfection efficiency and ability to grow in suspension cultures, which is ideal for continuous manufacturing. In contrast, AAV production often requires more complex handling and co-transfection with multiple plasmids, which could add complexity to the continuous manufacturing process.
Purification and Process Efficiency
Different viral vectors present unique challenges for purification. The choice of viral vector affects the ease and efficiency of separating the virus from host cell debris, residual plasmid DNA, and other impurities. For continuous manufacturing, a viral vector with properties that allow for easier or more efficient purification is critical. AAV, for example, requires careful filtration and chromatography steps due to its small particle size and heterogeneity, whereas LV vectors may be more readily purified through simple filtration or centrifugation methods.
The design of the purification process is critical for maintaining high product quality while minimizing processing time. In continuous systems, where viral vector production must remain consistent, purification steps must be both efficient and scalable. Poor purification can lead to contamination or loss of product, which ultimately affects the yield and safety of the final product.
Safety and Regulatory Considerations
Safety is paramount in any manufacturing process, particularly with gene therapy products that may be administered to humans. The viral vector selected must meet strict safety standards, including the absence of replication-competent virus and low levels of residual DNA or other contaminants. In continuous manufacturing, the selection of viral vectors with inherently safer profiles—such as those that are replication-deficient or have been engineered to reduce immunogenicity—helps mitigate potential risks.
For instance, when selecting adenoviral vectors for vaccine production, it’s crucial to avoid wild-type AVs, which could cause adverse effects. Similarly, lentiviral vectors are typically engineered to be self-inactivating to reduce the risk of insertional mutagenesis in target cells.
Process Control and Monitoring
Continuous manufacturing requires a higher level of process control and monitoring than batch-based systems. The selected viral vector must be able to tolerate and function under conditions of continuous flow, nutrient availability, and temperature fluctuations. Additionally, real-time monitoring of CPPs such as viral titer, host cell viability, and product quality is crucial. The viral vector choice can influence the type of sensors or analytic technologies needed for process monitoring.
For example, AAV vectors often require precise control of transfection parameters, and monitoring their titers in real time can be more challenging compared to other vectors. However, recent advances in analytics and inline process monitoring equipment have made it easier to track these vectors throughout the continuous manufacturing process.
Cost and Economic Considerations
Continuous manufacturing processes aim to reduce overall production costs, and viral vector selection directly impacts the economic viability of these systems. Vectors that require fewer raw materials, simpler purification steps, or more efficient cell lines may reduce costs and increase the profitability of manufacturing. Conversely, vectors that demand complex reagents, cofactors, or multistep purification methods can lead to higher operational costs. In some cases, a viral vector with higher complexity, like AAV, might be more expensive to produce than a simpler vector, such as LV. Thus, balancing the desired therapeutic outcome with production costs is a key consideration in the choice of viral vector for continuous manufacturing systems.
AVs have a packaging capacity of roughly 8.5 kilobases (kb)—a unit of measurement used to help designate the length of DNA or RNA. One kb is equal to 1,000 bases, high levels of protein expression, and transient gene expression. The onset of expression can occur as early as 16–24 hours after infection. The high immune response from the target cells is the main limitation of adenoviral systems. Despite this, they are still widely used in research due to their highly efficient transduction of most tissue.
AAVs have a packaging capacity of roughly 4.5 kb, relatively low levels of protein expression, and the potential for long-lasting gene expression. The tropism (the turning of all or part of an organism in a particular direction in response to an external stimulus) of AAV can also be increased via different serotypes. The primary disadvantage of AAV is its smaller packaging size for gene of interest, as well as a much later onset of expression (2–7 days for in vitro and 3–21 days for in vivo). However, this delivery system triggers very low levels of immune response.
Batch-Based Manufacturing: Lessons Learned
Protein production is often less complex than viral vector manufacturing. The batch production of viral vectors primarily relies on transient transfection of human embryonic kidney HEK293 cells using multiple plasmids. However, this method often results in low viral titers. Adherent cell culture is the most common approach, but there is a growing shift toward suspension-based processes. This process typically yields lower cell densities compared to those seen in the production of recombinant proteins and monoclonal antibodies (mAbs) AAVs. Although they are more stable than LVs, they face challenges during transfection.
This step often leads to the generation of a significant proportion of “empty” or “partial” viral capsids that lack the full gene of interest. In contrast, LV vectors are more vulnerable to degradation from mechanical stress, pH fluctuations, and temperature changes, which can result in low recovery during downstream purification. Viral vectors involve multiple proteins forming a complex three-dimensional structure that encapsulates DNA. Additionally, viral vectors tend to be more toxic to the cells used for their production than proteins.
The interest in viral vector research and development has steadily increased over the years, but production processes have been given only minor attention.
![Figure 4: Initiate quality risk management (QRM) process [5].](/sites/default/files/2025-05/0525_PE_MJ_Dream_05.jpg)
Risk Mitigation
In the production of certain biologic products, particularly viral vectors for gene therapy or vaccines, it is essential to operate within a biosafety level (BSL) environment. BSL guidelines are designed to protect both workers and the broader community from potential risks associated with handling biologics that may contain infectious agents or hazardous materials. This differs significantly from traditional Good Laboratory Practice systems used in mAb production, where the primary concern is generally the production of therapeutic proteins in controlled, noninfectious environments.
Hazards in Viral Vector Production vs. mAb Production
Unlike traditional mAb production—where the key hazards are often related to chemical exposure, cross-contamination, or equipment failure—viral vector production poses additional biological risks due to the inherent nature of the vectors themselves. Viral vectors, such as LVs or AAVs, may carry potential risks of unintended transmission or replication, especially in the case of replication-competent vectors. There is also a risk of unanticipated genetic modification or insertional mutagenesis when these vectors are used in gene therapy, which could potentially lead to harmful effects in recipients. Moreover, viral vectors could cause immunogenic responses or have harmful effects on the workers handling them, depending on the vector’s design and the safety measures taken.
In contrast, mAb production primarily involves cultured cells—e.g., Chinese hamster ovary (CHO) cells—that produce therapeutic antibodies. Although microbial contamination is always a risk, the primary hazards are related to chemical handling—such as reagents for cell culture, purification, or formulation—and the possible risk of product contamination. As such, mAb production can typically be done within a less restrictive laboratory or production environment when compared to viral vector production, which mandates a higher level of containment to mitigate the additional biological risks.
By producing viral vectors in a BSL facility, we can mitigate these risks. This ensures that the production process is both safe and compliant with regulatory requirements. These measures are vital to ensure the safety of production workers and the public, while also guaranteeing that the therapeutic products are of the highest quality and safety.
Virus Production and Handling
Dedicated top-tier virus production and handling facilities are required to ensure the highest standards of quality and safety. These facilities are equipped with BSL-2, BSL-2+, and BSL-3 laboratories, allowing manufacturers to manage even the most pathogenic viruses with unparalleled precision (see Figures 3 and 4.5, 6
![Figure 5: Overview of a typical QRM process [5].](/sites/default/files/2025-05/0525_PE_MJ_Dream_06.jpg)
Comprehensive biosafety levels
BSL-2, BSL-2+, and BSL-3 laboratories enable the safe production and handling of a wide range of viruses, including high-potency and high-risk pathogens. This ensures that a stringent requirement of diverse applications, from gene therapy to vaccine production, is achievable.
Continuous manufacturing processes aim to reduce overall production costs, and viral vector selection directly impacts the economic viability of these systems.
Rigorous quality control
Maintaining the highest standards of safety and efficacy and implementing stringent quality control measures throughout the production process ensures that viral products are reliable and meet all regulatory requirements. When performing quality risk management (QRM) for the continuous manufacturing process of AAV vectors, it’s crucial to evaluate several factors that could impact product quality, safety, and consistency. QRM is a structured approach to identifying, assessing, and mitigating risks throughout the manufacturing process. The following is a detailed outline of what to look for during the QRM of AAV continuous manufacturing:
- CPPs: Identify and monitor critical variables such as temperature, pH, dissolved oxygen, and glucose concentration (these can directly impact cell growth), AAV production, and viral vector quality.
- Critical quality attributes (CQAs): Establish the CQAs for AAV vectors, such as viral titer, purity (e.g., absence of host cell proteins or plasmid DNA), infectivity, and integrity of the viral genome.
- Scalability and robustness: Evaluate the scalability of the process. CBM requires a stable performance at a large scale, so ensuring that process parameters remain consistent as the scale increases is essential.
A typical manufacturing run of an AAV-vector therapy using high-yield cell lines and large-capacity bioreactors might only produce approximately 10 doses of a systemic gene therapy from a single batch, at a manufacturing cost of nearly US$100,000 per dose (or, about US$4.25 million in retail).
The Risk-Based Approach
The risk-based approach is applicable to continuous viral vector manufacturing. The quality, safety, and efficacy attributes of continuous viral vector manufacture and compliance with GMP should be ensured. This should be regardless of whether viral vectors are developed in a hospital, academic, or industrial setting (see Figure 5).
Manufacturers are responsible for the quality of the viral vector they produce. The risk-based approach permits the manufacturer to design the organizational, technical, and structural measures that are put in place to comply with GMP and thus to ensure quality according to the specific risks of the product and the manufacturing process. Although the risk-based approach brings flexibility, it also implies the manufacturer is responsible for implementing control and mitigation measures necessary to address the specific risks of the product and manufacturing process.
The quality risks associated with CBM production of viral vectors are highly dependent on the biological characteristics and origin of the cells/tissues, or the biological characteristics of the vectors. This includes, for example, replication competence or reverse transcription and transgenes, the level and characteristics of the expressed protein for gene therapy products, or the properties of other noncellular components (raw materials, matrices), and the manufacturing process.
When identifying the control/mitigation measures that are most appropriate in each case, the manufacturer should consider all potential risks related to the product or the manufacturing process based on the available information. This includes an assessment of the potential implications for the quality, safety, and efficacy of the product, as well as other related risks to human health or to the environment. When new information emerges that may affect risks, an assessment should be made to determine whether the control strategy (i.e., the totality of the control and mitigation measures applied) continues to be adequate.
The evaluation of the risks and the effectiveness of the control/mitigation measures should be based on current scientific knowledge and the accumulated experience. This risk evaluation is linked to the safety of the patients.
Cost Improvements
Many viral vector gene therapies require administering large quantities of viral particles to patients, particularly for systemic disease treatments. The large doses required for some gene therapies are challenging and expensive to manufacture.7 A typical manufacturing run of an AAV-vector therapy using high-yield cell lines and large-capacity bioreactors might only produce approximately 10 doses of a systemic gene therapy from a single batch, at a manufacturing cost of nearly US$100,000 per dose (or, about US$4.25 million in retail). Although these costs will gradually decrease as gene therapies begin to reach clinical and commercial scales, any technological advance that reduces the required dose would bring immediate benefit, as a 10-fold reduction in dose might also bring about a 10-fold reduction in costs.
Key business case drivers for viral vector manufacturing will be the realization of reducing manufacturing costs and increasing process optimization in the manufacture of viral especially. These two drivers can be realized to an identified level, making the business case for implementing continuous manufacturing over a traditional batch-fed mAb process platform.
The four basic approaches to the decision process for moving to a continuous platform are:
- Cost-of-goods improvement: This is the most common methodology implemented.
- Net present cost (NPC): NPC is comparable to net present value (NPV) but without a revenue component. This was introduced approximately 10 years ago.
- NPV methodology using a weighted average cost of capital: In this method, you would set a product transfer price for the baseline process that gives a NPV of zero.
- Portfolio management: This is the most sophisticated analysis methodology. It considers the uncertainty across an entire portfolio of products being considered.
Many manufacturing processes are initially developed in a batch-driven mode of operations, and subsequently “transferred” to a continuous platform. There is evidence that continuous manufacturing for traditional mAbs can enhance some unit operations in a batch-based platform. But overcoming some of the properties of the virus and the challenges around transient transfection make the move to continuous platforms more challenging.
Switching from batch-based to continuous manufacturing can lower manufacturing costs by enabling simultaneous upstream and downstream operations, reducing downtime, and increasing productivity at several-fold smaller scale. This is achieved using a range of technologies including perfusion bioreactors, single pass filtration modules, and multicolumn chromatography systems.
Contamination Control and Closed Systems
The majority of ATMPs and viral vector products cannot be terminally sterilized. In such cases, the manufacturing process should be conducted aseptically (i.e., under conditions that prevent microbial contamination). Manufacturing activities that may expose the product to a risk of contamination should be done in an area of appropriate environmental cleanliness level (i.e., production in a closed system, isolator, or positive pressure isolators). A background clean area of grade D is acceptable.
Isolators should be introduced only after appropriate validation. Validation should consider all critical factors of isolator technology. This includes, for example, the quality of the air inside and outside the isolator (background), disinfection regime of the isolator, the transfer process, and the isolator’s integrity. Monitoring should be carried out routinely and should include frequent leak testing of the isolator and glove/sleeve system. The transfer of materials in and out of the isolator is one of the greatest potential sources of contamination and as such, appropriate control measures should be put in place. When materials are added or withdrawn from the closed system without an aseptic connection (e.g., use of sterile connectors or use of filters), the system can no longer be considered closed. 8
In exceptional circumstances (such as the manufacturing of the many ATMPs where viral vectors are critical to chimeric antigen receptor (CAR) T cell therapies) it is not possible to move the production to an outside cleanroom because the time between the donation of starting materials and administration of the product is very short, and the patient is also in the operating theater waiting for administration of the product. Closed systems may be placed in a controlled but nonclassified environment. Operating conditions where manufacturing activity takes place should be adequate and sufficient to ensure quality and safety of the product.
The product should not be exposed to the environment (e.g., supporting data from leak testing and pressure check of the equipment). It should be demonstrated that the clinical benefit to the patient outweighs risks linked to the absence of a classified background.9 A detailed discussion on this topic of system closure can be found in the ISPE Baseline Guide Vol 6: Biopharmaceutical Manufacturing Facilities (Third Edition).7
Conclusion
The objective of moving large molecule drug substance manufacturing of viral vectors into a continuous operational platform focuses on improving manufacturing efficiency, effectiveness, and cost-of-goods improvements. These are all areas of interest for the gene therapy manufacturing companies that are looking for efficiency in improving process operations and utilization, improved control strategy, and a tool that will enhance production of key components of gene therapy products as demand for viral vector products increases.
The implementation of continuous manufacturing for viral vectors is being driven by the need for scalable, cost-effective, and efficient production to meet the increasing demand for viral vectors used in gene therapies and vaccines. Continuous manufacturing offers significant advantages in terms of productivity, process control, quality, and sustainability. It is a key enabler in advancing biopharmaceutical manufacturing for these high-demand products.


