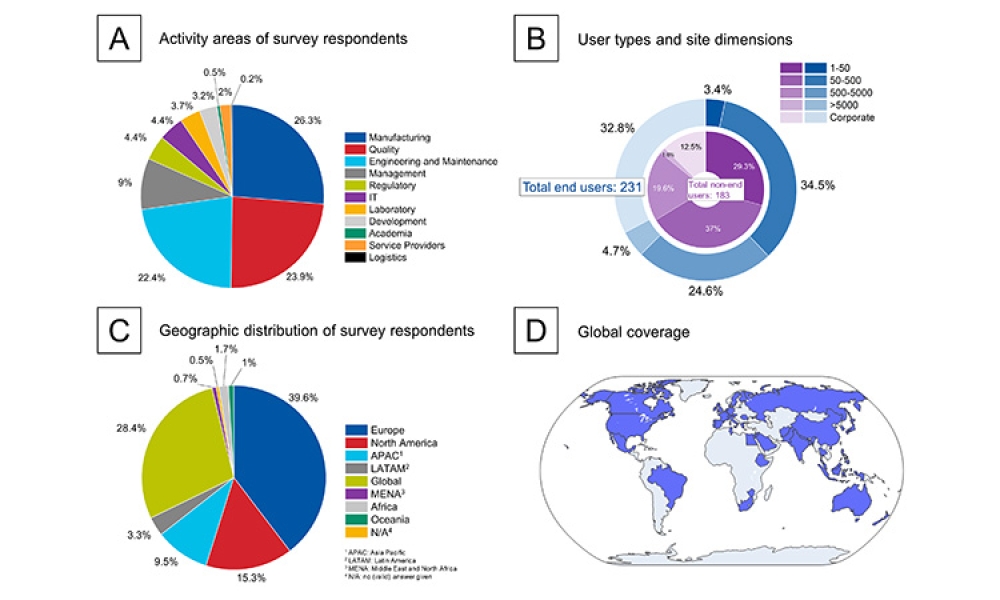
2022 ISPE Aseptic Conference: Regulatory Panel Discusses ATMPs, Data Integrity, Inspections, & More

The 2022 ISPE Aseptic Conference Regulatory Panel session on 15 March was as always one of the highlights of the conference, and this year’s discussion with regulators followed in that tradition.
Highlights from the panel discussion are presented here.
Panel Participants
Panelists:






Moderators:

What’s the direction you see for ATMP manufacturing regulation? Currently its practice is looser than cGMP requirements, I believe. Will the requirement for ATMPs be similar to c-GMP?
The future direction for ATMP production is so exciting. The science that is developing has such tremendous potential for clinical outcomes. Because it is such a rapidly evolving field, I see a need for the industry and the regulators to be very well connected and engaged as much as possible. From a PIC/S perspective, we do not see the GMPs for ATMPs to be looser compared to requirements for other products. Within the PIC/S framework, ATMPs are subject to the same GMP provisions as other products as noted in the PIC/S GMP guide, part 1 and part 2. Many of the PIC/S annexes also apply to ATMPs, including Annex 1 for sterile products, and Annex 2a that was written for ATMPs. It was written in a way to reflect the potential for rapid advancements and quick changes in the field. There's going to be batches that will be patient specific, and these are smaller scale production activities that may require a regulatory framework that enables multiple batches to be produced in the same area. What controls need to be considered when patient-specific manufacturing has multiple products and different viral vectors in the same room? The Annex helps consider that there's also added or elevated GMP requirements. When we talk about human cells and tissues, we have different needs for traceability of the starting materials, the raw materials, tracking for longer periods of time and recordkeeping requirements, including processes for look backs. If health information arises from donor information, there are also risk-reducing actions that can be considered as alternative to recalls. In the case of an autologous ATMP, what is better: a recall or other options where patient treatment options and the risk to patient treatment options might present a higher risk than a potential recall product?
What data integrity issues are inspections uncovering in aseptic manufacturing operations?
One of the examples that I would like to give here is incubation of blank or clean plates for environmental monitoring to give the impression that the results were good. These might be plates that were never placed out for sampling operations, or the results were manipulated. Another example would be precompletion of batch records. We do discover these in inspections, unfortunately, and it raises a lot of questions around the culture of a company that would support this type of activity. This directly leads to the conversation about the authenticity of all data and issues with data integrity as a whole.
I'd like to concur that data integrity issues and poor data governance can be found in any sector of pharmaceutical manufacturing. It can be in production or quality control. It can be as a result of fraud, intentional, or simply rising from poor practice. And I'd like to make industry aware of a PIC/S document that's intended to guide inspectors on good data governance practices. This is the PIC/S Guide to Good Practices for Data Management and Integrity in Regulated GMP Environments. It's worth noting that our host for this event, ISPE, also has a number of guides on data integrity that can be a source of information to facilitate compliance, as do many other organizations.
Data integrity citations in fiscal year 2021 dropped significantly, and the lack of physical inspection is widely considered part of the reason. How will this be addressed going forward?
While we conducted fewer inspections, the inspections that we did focus on were those that the agency considered mission critical. We have resumed domestic inspections. We are gearing up and are planning to resume our prioritized surveillance inspections. We have for some time put as a priority those mission critical activities that I mentioned before, but also increasing in the number of preapproval or application-based inspections, as well as those that were OAI [official action indicated] or for cause compliance follow up inspections. It is presumed that sometime in June we will be getting closer to a normal cadence of inspections. Let me also add that we have been putting additional resources into our India office. We believe that these measures will certainly help us to get back up into a normal cadence with conducting foreign inspections, and this will get us back to finding these critical deviations where data integrity issues may occur. Certainly we recognize that the on-site inspection still represents the best outcome for understanding the compliance practices that occur at a firm.
I wanted to mention that we encourage you also to self-report any data integrity issues that you find and remaining transparent with regulators. We can work together as you identify, assess, and address any of these data integrity issues. If we identify the integrity issues during an inspection, then we're put into a reactive mode rather than a proactive mode. It's also very awkward if we happen to stumble upon the data integrity issues during the inspection, especially if you're already aware of them. Although you might not mean to hide them, it might appear to be that way. So, maintain that transparency and self-report where possible.
Should regulatory bodies set acceptable residual hydrogen peroxide levels specifically for biologic drug fills? And if so, at what ppb levels?
Generally speaking, our regulations are set very general. We don't want to be too dogmatic about what we require. My first response when I saw this was if we set a specification, somebody is going to be unhappy with whatever it is. I think with biological products, as they're so varied, it would be hard to set one level to fit all the different products. I think the bottom line is you need to demonstrate what works for your product and we'll work with that.
What was the FDA's approach to approving new isolation technology, for example, of VANRX for commercial manufacturing? More specifically, how did you approach compliance with the current regulations and requirements?
I won’t be discussing specifics related to the vendor referenced in the question, but I can speak about robotic isolators. CGMPs are open to a lot of technological options, and they leave the field wide open for beneficial innovations to be implemented within the context of GMP. The FDA guidance for industry does not overprescribe and allows this latitude. While GMPs do not specify a given technology that has to be used, they do provide the quality systems framework to implement technologies such as robotic isolators. And of course, we're very excited about new technologies that meet the GMP imperatives of robust manufacturing and reliable quality We understand that the landscape is sometimes a bit ambiguous for new technology. So, through our emerging technology program, we've encouraged dialogue on novel technologies by providing a real opportunity for engagement with companies on these trailblazing efforts, such as the robotic isolators. With that said, it's still aseptic processing, which is one of the most unforgiving processes in the industry. The primary failure mode that we're particularly trying to exclude by using isolators and robotics is the personnel element of it. Sterility failures are uncommon in isolators because of the greatly reduced hazard from aseptic operator interventions, and robotics will further cement that huge advantage. However, any aseptic process has multiple failure modes. For example, when advanced technologies like isolators have had an infrequent sterility or media fill failure, it was typically caused by mechanical issues including but not limited to CIP/SIP drain piping back-siphonage, non-sterile silicon, and pump breaches and leaks. The drain issues have led to Bacillus, spp. contamination in multiple instances. . These and other failures were not caused by aseptic technique. Some of these were detected by media fills. Others were detected by the crucial final QC sterility test. These aseptic failure modes are universal, including for isolators. These findings underscore the public health importance of the requirement to test any aseptically processed batch before QA can determine the suitability of any batch for distribution to patients.
So it is important to have the right management control and design approach including the right detection systems in place. This includes media fills and sterility testing, but also ongoing environmental monitoring and other detection systems. So as we move to the next level of assurance with robotic isolators, we should remain vigilant about overseeing ongoing process control. We should always remember that complacency is the enemy of quality. Fundamental failure modes of any process remain a universal truth, even though they're greatly mitigated if you install the right robotic isolator. If designed and controlled right, it will lead to better, sustainable assurance of supply and quality every day. But if you become complacent, obviously you may be in a reactive or crisis mode later, even with a robotic isolator.
There is a lot of good engineering out there. But what I am finding are some fundamental issues in the design phase where the people engaged in environmental monitoring are not part of the discussion. Because of an automated system and robotics, that does not preclude you from an environmental monitoring program. What is it that I want to capture with an environmental monitoring program? That is regardless of whether you have a totally robotic system, an automated, semi-automated system, an isolator, or a RABS. With an automated system, you can design something where you don't have to monitor as frequently or only in certain locations. But what I'm finding to be true with automated systems and robotic systems is that some of those fundamentals are not considered during the design of these systems, such that I've had companies come back and tell me they have to go back to the designers of the equipment to address some issues; as an example, with monitoring for the presence of non-viable particles.
And we want to see companies complete the move to isolators as we know that hugely mitigates risks. At least 90% of contamination (in data from industry surveys) comes from people as the direct source. But we of course must remain aware that the remaining residual risk of mechanical failure is still there. Some facilities have been found to not fully understand their failure modes. To reinforce Tom’s point, they don't always have the right people at the table when designing and creating the control and risk mitigation approach for RABS and isolators, including some that use robotics. With that said, they are making a huge difference and there are incentives in our guidance. Both the aseptic guidance and our cGMP Q&A that say you could generally justify doing less media fils for isolators specifically -- just two per year generally instead of two per shift per year for non-isolated lines. And I think that public safety, efficiency, and these pro-modernization policies are why the industry has moved so much to isolators, because they make good quality and good business sense. Just do it right, like Tom said.
What is the expectation on environmental monitoring in an automated line? Do we need to conduct all settle plates, contact plates, and active sampling by robots or manually?
What is it that you want to accomplish by monitoring your facility? For an automated system, perhaps we can evaluate whether we need to perform the full litany of different samples, whether it is a passive sample, active sample, or others, when compared to a non-automated line and I'm going to use a RABS as an example. Well, it's not automated, there's a lot of manual activity and because of many design features of a RABS, you're going to do some monitoring. And that's perfectly understandable, if you remove all those activities where you don't have that person interaction and it's totally automated, or you do have some automation where you can reduce the presence of that personnel activity. Your monitoring strategy must be risk-based and must achieve the goal of proper product control. If you present your scientific reasoning for your monitoring, including the data to support your scientific conclusions why your monitoring program is suitable, then that can be successfully presented to FDA. And that is regardless of whether your filling system is automated or not.
Too often, aseptic processing lines are de facto semi-automated as we still see a lot of interventions in routine operation. An isolator can be considered an essentially closed system, whether it is fully physically closed or it has mouseholes for transfers. Obviously, it is not hermetically sealed. But for isolators, we have supported surface monitoring at the end of a campaign instead of every day and microbiological swabbing of gloves can generally be at the end of a campaign as well. Multiple air samples should continue each day for isolators, of course.
For RABS, the numbers of environmental monitoring samples are like that used for traditional lines and it is important to justify locations. More intensive microbial monitoring of the inside of the RABS and its of surrounding rooms is needed versus that in an isolator. RABS are opened at times, and they also only rarely have automated decontamination. I think you all realize these and others differences between robustness of a RABS environment, which is still vulnerable to so many variables, versus closing off the ISO 5 environment in an isolator. The isolator creates more of an internal micro-system that you could focus on maintaining and do much less microbial monitoring outside the isolator and can also allow for stepping back from daily surface monitoring in most isolators. And you can further mitigate risks by performing the active air monitoring using remote monitoring technology throughout the campaign to count and facilitate transport to identify the isolated microbes. Isolators have so many efficiencies in that respect and that's where we focus. They also have cost savings regarding a smaller footprint and less gowning, in addition to less environmental sampling.




If filters are 100% integrity tested prior to integration into a non-sterile single use system, is there a risk that the single use irradiation process will create defects in the membrane that would require PUPSIT (post-sterilization, pre-use integrity testing)?
Rather than focusing on PUPSIT, I'm going to focus on the specific question of risk that a radiation process has on a filter when you integrate it into a single-use system. This places the responsibility back into the manufacturer’s decision-making processes. Each manufacturer needs to consider elements within this as part of their quality risk management framework, building on the knowledge of the process, knowledge of the impact of any irradiation process, and when working or obtaining such products from suppliers. This could be important from a supplier qualification perspective and in the end the responsibilities lie with the manufacturer to ensure there's good science and appropriate processes.
Do you expect or prefer incubators for aseptic ATMP manufacturing to be decontaminated? It's usually one of the riskiest parts of the ATMP aseptic manufacturing.
It looks like we are talking about an incubator that is used for the growth of a product at an early stage of the processing. Since you think that this is the riskiest part of the process, then I would hope you had decontaminated the unit prior to initiating the manufacturing. If it is an isolator-type CO2 incubator, then we would also expect decontamination of that as part of the process. If it is an open system that is being used inside the incubator, we would hope that there is some sort of filters on the containers that are being used to grow the product or the cells. We would expect any part of the process used for aseptic processing to be decontaminated.
I'm going to draw us back to the PIC/S Annex 2, which is for manufacturing of ATMPs. In that Annex, we use specific terminology that states cleaning and disinfection procedures for incubators. Now maybe decontamination might be a more appropriate term. There are complexities to this question: What stage in the processing are we talking about? How is the incubator being used? Are there design considerations? Are there process considerations? Are separate incubators being used for different organisms or cells? Is this an incubator system where more than one organism or cell type could be located? What are the control steps that are in place? How are they sealed? What's the surface decontamination? How are containers segregated within an incubator system? And the list keeps going on. Are there manufacturing activities involving infectious viral vectors, oncolytic viruses, or replication vectors? Once again, it all turns back to the ATMP manufacturer to apply good quality risk management in their processes in developing and pulling together a contamination control strategy and taking the most appropriate approach with all the factors under consideration.
What kind of usage are you expecting for enzyme indicators for H2O2 decontamination? Do you see any possibilities that enzyme indicators replace biological indicators?
The way I read the question is: will enzyme indicators or chemical indicators replace biological indicators? Those two indicators provide you with very different data. A biological indicator will give you quantitative data. You will be using a biological indicator to determine the reduction of a known number of spores, when applying H2O2, or peracetic acid, or any other chemical as your decontamination system. This will give you some very good data: with a certain concentration of H2O2 over a certain period of time, under certain conditions and in a certain location, you can demonstrate that you have an x-log reduction of your known number of organisms. With a chemical or enzyme indicator, it is just qualitative data that you are generating. In my opinion, a chemical indicator is not going to replace the data that you're going to get from a biological indicator.
I see a possibility for a hybrid approach where enzyme indicators are used to gain significant information and help with qualification. The information available and the understanding of distribution and dwell time of the H2O2 in a particular area as it moves turbulent around an isolator will be extremely valuable. If you want to know that it gets to those tough to reach spots through turbulent flow and achieves a consistent kill, this can certainly improve the quality and quantity of data in qualification initially and on an ongoing basis. So this approach could augment biological indicator data and help to fortify isolator qualification and validation. I know papers have been published on this topic.
Air flow pattern evaluation is done to visualize the airflow in confined spaces, and there is going to be some level of turbulences. If you have turbulences, you can use chemical indicators to identify areas where there is less contact of H2O2with a surface, and that area could be the source of cross-contamination to other areas due to the airflow. This will be part of the development of your system for decontamination, but also for the development of your environmental monitoring program.
Considering the significant investment in manufacturing capabilities seen in the past few years, can the panel speak to the vision to keep up? What should the industry do to aid the regulatory ramp up?
The industry has greatly driven isolator upgrades and barrier upgrades and we have supported you with our encouragement, participation in this annual form, and clarifications on numerous technical questions. Bob and I have been part of this for the last 25 years, and we are very proud of the fact that we have helped to demystify isolator and barrier technology for the industry and have given you a consistent message on it. The case studies presented at this conference, and industry surveys initiated by ISPE have helped a lot. ISPE deserves great credit for that. You brought quantitative data to explain the business cases for isolators and barrier systems, and there have been presentations and book chapters on it. I think industry needs to continue to improve capabilities and to keep advancing this vision, to share lessons learned, as it is in everyone’s interest to advance public safety for patients worldwide through use of today’s more capable technologies. We as regulators continue to be trained well in the centers and ORA on modern technologies, and have adapted regulatory approaches. We have the very broad GMP construct since 1978, which allows for these innovations. Annex 1 calls it contamination control strategy, which of course is ultimately a contamination prevention strategy when it comes to the product. Let’s continue working together in getting to the next level of innovation. FDA can provide feedback through multiple appropriate mechanisms, including the Emerging Technologies Team, depending on the status of your site and its future plans.
Can you give some background to the recent draft guidance for industry from FDA on the visual inspection that came out recently?
The draft was issued in December 2021. This is an important area for parenterals. We are filling that void where USP published particulate testing monographs but does not define the expectations of cGMPs. We are talking about CAPAs when improvements in operations are needed. We discuss intrinsic and foreign particles, and how they can jeopardize patient safety. We discuss a holistic risk-based approach to improve the performance of the industry and reduce the risk of introducing hazardous visible particles into the drug formulation in parenteral manufacturing.
The leaked version of Annex 1 (version 13.1) had guidance values replaced by minimum requirements. Is this going to also be in the final version?
There was the change to incorporate content that says were specific limits or frequencies or ranges are written, they should be considered as a minimum. And there's a reason behind this. They're stated due to historical regulatory experiences of issues that have been identified and have impacted on the safety of patients. I've already mentioned this during the presentation, but there is going to be flexibility to incorporate QRM (quality risk management) and good decision making where some of these minimum requirements are specified. A good example is leak testing of autoclaves, which has a minimum frequency recommended as once week, but if you aren't going to use that autoclave on a weekly basis, there's good justification and rationale to present other time periods.
Not interrupting first air in RABS and isolators is a goal in Annex 1. In practice, we know that this will not always be possible. How should this be approached?
Interrupting first air should be avoided at all costs. Airflow pattern evaluation and visualization will help you in this regard. What we strongly encourage is that you include the operators and those performing environmental monitoring in the design of your RABS or isolators. We all agree that first air should never be disrupted. A well thought out design should include considerations to prevent disruption of first air. In the event that first air is inadvertently interrupted, it should lead to a documented evaluation of the contamination hazard posed to the aseptic operation.
Eudralex Volume 4 has provisions for investigational ATMP guidelines. Is there an equivalent guidance document from US FDA?
There is a guidance document, cGMP for phase 1 Investigational Drug Products, which was published in July 2008. This was for CDER and CBER (maybe also for CVM). We expect adherence to cGMPs under 501 A and B of the Food, Drugs and Cosmetics Act for products used in patients at all clinical phases, but the full CGMP regulations at 21 CFR 211 become more applicable starting at Phase 2.
What is your stance on continuous process verification and using it to replace time based, for example, annual or biannual requalification?
Qualification and continuous process verification (which is a validation term) are conflated in this question. They are two different things. You qualify equipment and you validate processes. CPV (continuous process verification) is about process verification on a continual basis. Let’s use continuous manufacturing as an example. If you have a continuous process stream in drug substance manufacturing, or maybe in a tableting or capsule filling process, you can use validated on-line measurements like RAMAN or NIR spectroscopy as a feedback/feed forward mechanism that allows diversion of non-conforming material and acceptance of material that meets the specifications. In sterile operations, there really is not a model for continuous verification because you cannot continuously verify the sterility attribute. You can do it with weight uniformity of vials on a line, however. We see a lot of 100% fill weigh checks, and kudos to the industry for advancing in this area. We strongly encourage that. There are places where this kind of concept can be applied in the validation and verification sense. But equipment qualification is a different animal, although a building block in the overall master validation plan. You have to make sure the equipment stays in a state of control to continue to support the state of validation of the process too, but that has its own considerations relating to predictive and preventive maintenance.
What role do you foresee single-use systems playing in the future, given the push towards sustainability and sustainable technology?
Climate change and the environment are part of so many important considerations that we need to consider as we work together in making sure that patients have continued access to quality medications that are safe and effective. It's something that we all need to engage in, regulators and industry, as we work towards sustainable approaches. We need to explore the environmental impact of single-use systems. But in doing so, we also need to weigh these with understanding of the impacts of the systems that we replace. What is the impact to generate high purity water or the heat that goes into our cleaning and sterilization processes, which offers a better overall risk benefit profile? What about the waste that's generated in single-use systems? Are there opportunities to leverage the waste such as waste to energy systems, or incorporating the waste into other products? But this brings up other considerations, such as biohazardous risks that could be present. So I think it's a very complex topic, and I think it's one that we all need to continue to work towards in terms of making a more sustainable future, how we can better protect the environment, and how we can contribute to addressing climate change.
We only have warehouses and we distribute drug products for the US market. What are the primary processes and systems an FDA inspection would focus on for this type of facility?
This is a question of basic material management. How do you control material coming in? What are the steps to ensure that the material is stored as recommended by the manufacturer? Are there any specific temperature conditions? A very important question is also: When product is being shipped and loaded for transportation, who owns the material at that point in time? Is it the customer? Then he is responsible for transportation. If you still own it until it reaches your customer, then you are responsible. It is all just basic good material management, making sure that the material is not compromised during storage or shipment.
What has COVID-19 taught us about inspections?
Alonza Cruse: We have to rely on our partners more than ever, and we need to continue to leverage and collaborate on those areas where we have a mutual and joint interest.
Thomas J. Arista: I can confirm what Alonza was saying. We as regulatory agencies around the world have learned to collaborate more. We might have respectful differences of opinion, but that is a positive thing.
Paul Gustafson: I think COVID has taught everybody, industry and regulators, so much about how to work together, so much on how to find new creative approaches, to address challenges that we're faced with and come up with strategies to move on very quickly and very effectively. And it really has enabled us to bring some lessons to advance and improve on in the future.
Robert Sausville: The pandemic has demonstrated how important it is that we all work together. We can get a lot of things done.
Alonza Cruse: It certainly has unleashed creative problem solving over the past two years and something that I think will certainly continue to launch us moving forward.
Rick Friedman: We learned the same lesson as industry: Communication and collaboration lead to greater efficiency and effectiveness.
Could FDA please comment on how they're going to work through the backlog of foreign inspections with so many open positions in the inspectorate?
While the pandemic postponed much of our surveillance work and activities, it really didn't stop the activities internally. Over the course of the past two years, we hired close to 80 investigators, and we had to adapt our training program in light of the public health emergency. We have been working to get staff up to speed as quickly as possible with regards to the training and other activities. As travel regulations are eased or lifted, we'll continue our risk-based, prioritized inspections. We are working with our regulatory partners across the globe to assist in mutual recognition. Those agreements will certainly help us in in dealing with some of the inspectional activities that we have going on. We even expanded beyond MRA [mutual recognition agreement] and really got creative. As many of you know, the MRA is for inspection work done in EU member countries and the work done in the US and we exchange that information. Now, we even sought to look at inspectional activity reports that were conducted by EU member countries outside of the EU working with PIC/S on mutual reliance. We certainly recognize that the inspection remains the gold standard in terms of what we do. I'll certainly mention now that while as regulators, we bear responsibility in terms of oversight aspects, the ultimate responsibility really lies within the manufacturers. And I think Brooke hinted at it when she emphasized the importance of the self -reporting, the importance of the self-auditing, and the importance of looking for problems and finding corrective actions prior to any oversight of any regulatory body or any auditor.
Is there a difficulty for FDA to rely on MRA? What can firms do to help the agency prioritize inspections as we emerge from COVID-19?
We have been working with our EU member partners and have asked them for inspections at specific sites, they have done the same for FDA to inspect US sites, and this will continue. We really have to rely on the institutional knowledge of our respective regulatory bodies to assist.
It's clear that regulators want to get back to inspections. Do you have any comments on learnings from COVID-19 and ways to modernize inspection approaches and a path forward with industry?
The answer to that question is still being evolved. We have engaged in the use of additional alternative tools, like request for records under section 704(a)(4) of the Federal Food, Drug, and Cosmetic Act [21 U.S.C. 374(a)(4)], which we have had since 2012. The pandemic gave us an opportunity to really expand on the use of that activity. It is something that we are going to continue throughout the use of the remote interactive evaluation live stream or teleconferencing. We've learned a lot over the past two years, and we are still learning about conditions under which it works best to use one alternative tool versus another alternative tool. How do we combine a records request with a physical inspection? How can we best use those tools to help us determine compliance within a respective firm? We see this as extensions of what we are doing, also post-COVID.
The FDA has resumed both domestic and foreign inspections. However, our company has noticed FDA is scheduling foreign surveillance inspections within MRA countries for no obvious reason. Could FDA perhaps elaborate?
When we are traveling internationally, then that is focused on mission critical pre-approval inspections. If we are conducting an inspection in an area and if there is another site that's within our risk model that we can readily do an inspection at that doesn't increase a quarantine period or doesn't add significantly to travel, we are looking to schedule that inspection and that's really just part of a good governance structure since we've already employed an inspector. That's one reason and that's probably the dominant reason why this is occurring. Sometimes the request that is being asked is beyond the scope of the inspectorate within an EU member country, and we will look to conduct that inspection ourselves, inviting the local inspectorate to join. These are two reasons why this might have happened.
Is FDA giving any consideration to using remote interactive evaluations introduced during the pandemic as a permanent substitute for facility inspection?
We have developed remote interactive evaluations as an alternative tool, and we will continue to refine it to assist us whenever travel is prohibited, even beyond COVID.
At the beginning of the panel session, Bob pointed out in his disclaimer that the discussion during the panel session is subject to 21 CFR 10.85 (k), which means that whatever was said represented the individuals’ opinions and their best advice, which reflected the FDA’s current thinking, but the advice is non-binding and cannot be held against FDA.
Disclaimer
Disclaimer: This article contains an abridged, unofficial summary of regulators‘ responses and discussion during a panel dialogue at an ISPE conference that has not been vetted by any agency or organization. The responses are an informal and brief synopsis of the panel’s views, and do not represent official guidance or policy of any agency or organization.
About the Author