A Proposal for a Comprehensive Quality Overall Summary

When working with the common technical dossier (CTD), the structure of Module 2 “follows the scope and outline of the Body of Data in Module 3,”

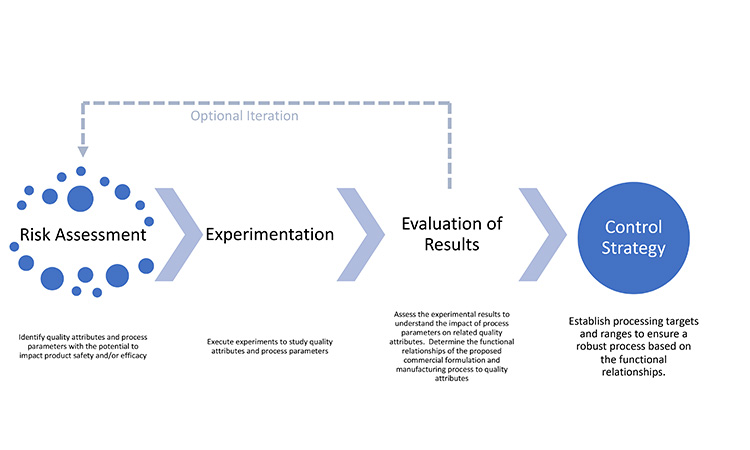
This assessment is performed for each CQA in turn, identifying parameters or variables that have the potential to affect those attributes. The results of the risk assessment are used to define and prioritize experiments. Results from experiments may confirm or modify preliminary risk assessments. Table 2 shows an example of a finalized risk assessment after experimentation, where items highlighted in yellow and red are described in the regulatory binding elements of the control strategy. Standardizing risk assessment summaries reported in applications will increase transparency and facilitate agreement between global regulatory authorities and industry on appropriate and acceptable control strategies.
Table 2: Example of a final risk assessment summary table.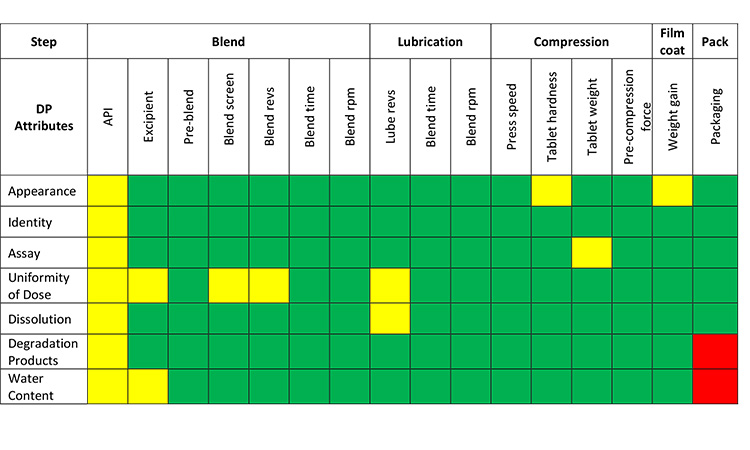
Key:
- Green: Parameters or material attributes have no relationship to a CQA. Non-critical controls are in place.
- Yellow: Parameters or material attributes have a relationship to a CQA. Critical controls (ECs) are in place.
- Red: Parameters or material attributes have a relationship to a CQA and an edge of failure has been identified. Critical controls (ECs) are in place.
The iterative cycle of risk assessment and experimentation can be considered complete when the relationships between parameters and CQAs are understood and take into consideration the broader context of the clinical significance of CQAs and the ability to control them during processing.13 Transparent communication of the relationship between CQAs, risks, and mitigation strategies will benefit the regulator assessing the application as well as the MAH assessing potential post-approval changes.
A summary of pharmaceutical development that presents the knowledge gained through the application of scientific approaches and quality risk management during the development of a product and its manufacturing process12 provides substantiation for the regulatory binding information that define a product control strategy. Provisions for additional detail may be established by hyperlinks to Module 3 within the CTD.
Presenting the Control Strategy
A control strategy consists of “a planned set of controls, derived from current product and process understanding that ensures process performance and product quality. The controls can include parameters and attributes related to drug substance and drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control.”12 Elements of a control strategy may be parametrically based or may be primarily focused on control of process outputs (e.g., attributes, measurements, responses).3 Control of a CPP and/or CMA is achieved by understanding the relationship between input and output variables in a manufacturing process, including material attributes, in-process controls and process conditions, and operating parameters. Every CPP (CMA and CQA) represents regulatory binding information.
While CMA has not been endorsed as an ICH term, it is being widely used to distinguish the variability associated with conditions in a process, known as process parameters vs. variability inherent in materials used in the process, known as material attributes. Regulatory binding information may also include non-CPPs and non-CMAs in addition to CQAs and CPPs. The distinction between critical and non-critical attributes and parameters will be necessary to ensure regulatory oversight of post-approval management is appropriately differentiated.
Specific controls can be established within the manufacturing process where the boundaries are defined by the inter-relationship between process parameters. Specification criteria are often established to control CMAs. Analytical methods are developed and adopted to evaluate specific materials—i.e., raw materials, intermediates, DS, formulation, and packaging components, DP—against pre-defined specification criteria to confirm control. While all controls are intended to assure quality product, finished product testing, by itself, does not constitute a control strategy.
The comprehensive control strategy can be visualized in the pictorial diagram shown in Figure 3. The backbone of the control strategy is made up of the CQAs shown as red boxes. Process parameters, material attributes, in process tests, release tests and GMP controls which are critical are represented by red ovals, while those that are non-critical are represented by blue ovals. The diagram shows that some elements of the control strategy have primary functional relationship to the CQAs, while others may have secondary, tertiary, or beyond. Further, it is shown that some CPPs may impact more than 1 CQA. For example, CPP15, which is the number of rotations used during lubrication blending, impacts CQA4 (content uniformity) and CQA5(dissolution). Other such examples are apparent in the diagram. It is envisioned that critical elements of the overall control strategy mapped in Panel B would be described in Module 2.
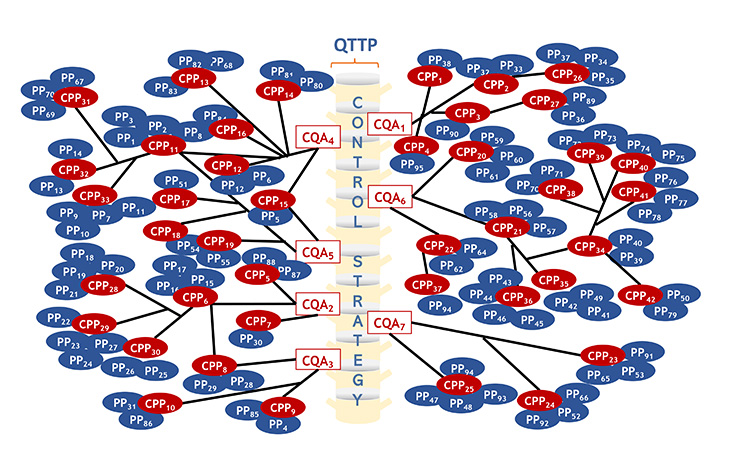
Panel A: Functional relationships among CQAs and various process parameters, material attributes, IPCs, tests and GMP Controls. The details underpinning this would be described in Module 3.
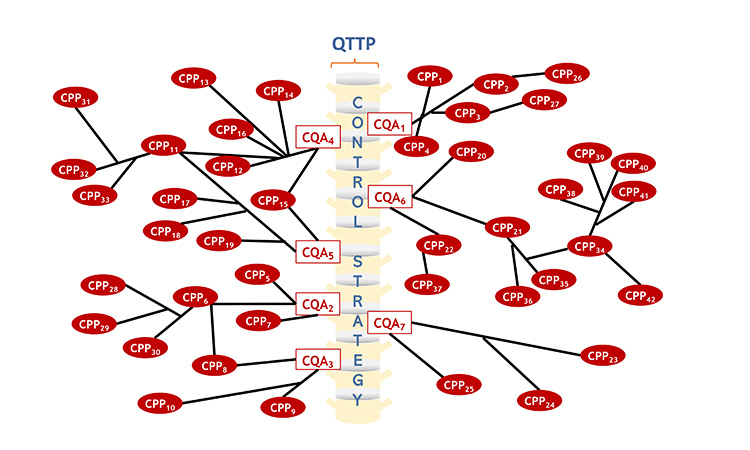
Panel B: Functional relationships among critical elements of the control strategy. The elements of the overall control strategy shown below would be described in Module 2.
About the Authors






Acknowledgements
The authors would like to acknowledge the contributions of the following colleagues, especially those involved with drafting the original PhRMA white paper and those involved with reviewing the current paper, including Dan Bollinger, Andrew Chang, Xavier Castell, Graham Cook, Frank Diana, Olivier Dirat, Jeff Ferguson, Georges France, Tom Garcia, Betsy Fritschell, John Groskoph, Nirdosh Jagota, Bob Kelly, John Lepore, Rick Lit, Stephen Mason, Moheb Nasr, Sarah Pope Miksinski, Ron Ogilvie, Ken Oh, Mark Rosolowsky, Greg Rullo, Tom Schultz, Steve Tyler, Timothy Watson, Jim Webb, and Diane Zezza.


