

A GAMP® Approach to Computerized System Life Cycle and IT Process Records

This article describes a practical and pragmatic approach to the management of computerized system life cycle and information technology (IT) process records. The objective is to effectively achieve and maintain compliant GxP-regulated systems that are fit for intended use, and to support patient safety, product quality, and data integrity.
Introduction and Overview
A GAMP® best practice approach is achieved by applying current good IT practice and modern software engineering techniques as well as principles supported by the effective use of commonly used standard software tools. Using practices that have been traditionally applied to GxP records, particularly paper records, can be inappropriate and unnecessary. Doing so may also potentially cause harm by:
- Increasing complexity and therefore risk
- Discouraging the use of widely used and well-understood standard tools, the application of modern methods and techniques, and effective communication between those involved
- Encouraging unnecessary duplication of information, cumbersome manual workarounds, and the development of custom, often homegrown solutions
Computerized system life cycle and IT process records managed with modern techniques and automated tools are not uncontrolled. They can be rigorously managed through access control, privilege management, security features, auditing and logging, enforced roles and responsibilities, segregation of duties, and using many other features and functions that are built into modern tools.
Robust, reliable, and effective GxP computerized systems that support product quality and patient safety require the application of effective modern methods and techniques that follow accepted software engineering principles and IT good practice. This includes processes such as version and configuration management, requirements management and traceability, testing, verification and defect prevention, and release management. In a modern software and IT environment, this can only be achieved by using suitable tools.
CAPA: corrective and preventive 1
DBMS: database management system1
Scope
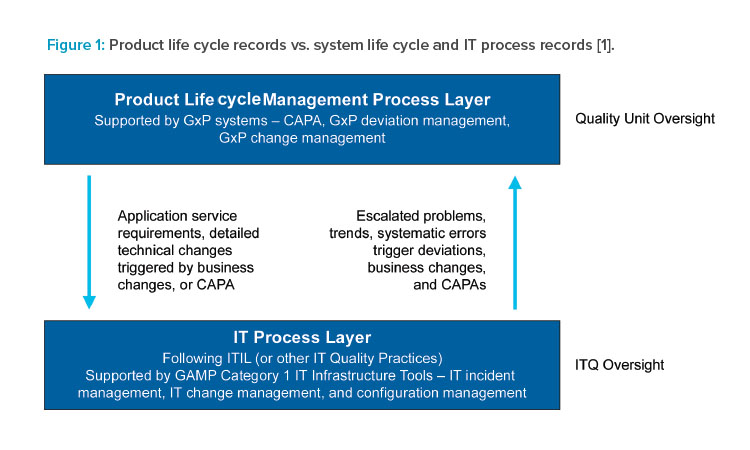
GAMP® guidance makes a valuable distinction between (1) GxP records supporting the medicinal product life cycle and required by predicate rules, and (2) the information, data, and artifacts that support computerized system life cycle and IT process records (see Figure 1). This distinction, applied with critical thinking, supports an approach that is effective in reliably delivering and maintaining systems that are fit for their intended use.
Examples of computerized system life cycle records include requirements, specifications, designs, test definitions, and test results. IT process records include those supporting incident, problem, capacity, performance, change, and configuration management processes. Such records are valuable to the organization and should be securely and effectively maintained as part of the IT quality management system, but they do not directly support the GxP medicinal product life cycle.
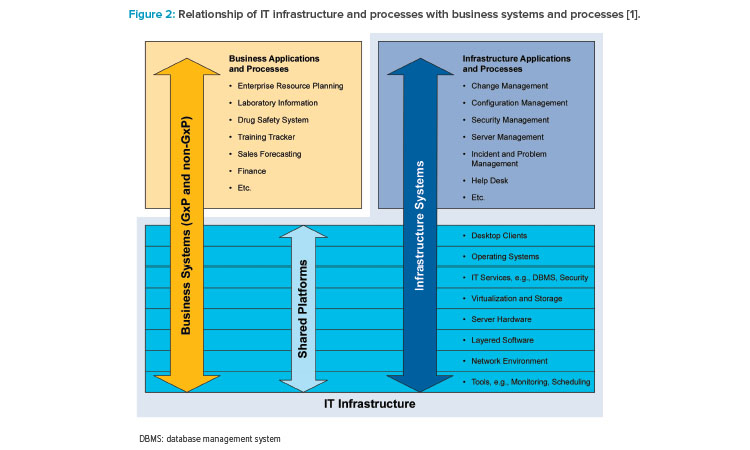
Records supporting system life cycle management, IT, and infrastructure process are typically maintained in GAMP Category 1 systems or tools, as shown in Figure 2. A modern best practice approach to the use of such tools is described in Appendix D9 – Software Tools of GAMP® 5 (Second Edition).
As noted by the US Food and Drug Administration (FDA), such records are kept for the benefit of the company, for a specific purpose, and to support business processes and objectives: “The work that you do should be valuable to the organization, right? You’re maintaining a record. You’re maintaining the activities, not necessarily because you need to demonstrate it to the agency or to any other auditor. You’re doing this work and maintaining this record because it becomes your source of truth for your organization down the road.”2
Examples of GxP records supporting the medicinal product life cycle include those for clinical trials, master production and control, batch production, calibration, cleaning, deviation and corrective and preventative actions, and pharmacovigilance. GxP records also include formal validation plans and reports, including those for process validation and analytical method validation, and data that directly supports a GxP activity (e.g., process performance qualification batch data used in commercial products). Such validation plans, reports, and data should be managed using GxP document and records management approaches.
Computerized system validation plans and reports would be regarded as GxP documents in the same way. Other life cycle deliverables should be managed by applying normal and easily achievable good documentation practices, or, if in a format other than a traditional paper or electronic document, be maintained securely in an appropriately managed and controlled tool or system.
US FDA Narrow Scope
As noted by the US FDA, inappropriate application of Part 11 requirements can lead to unnecessary controls and costs and can discourage innovation and technological advances without providing added benefit to the public health: “…concerns have been raised that some interpretations of the part 11 requirements would (1) unnecessarily restrict the use of electronic technology in a manner that is inconsistent with FDA’s stated intent in issuing the rule, (2) significantly increase the costs of compliance to an extent that was not contemplated at the time the rule was drafted, and (3) discourage innovation and technological advances without providing a significant public health benefit.”
Part 11 also addresses narrow interpretation of scope: “We understand that there is some confusion about the scope of Part 11. Some have understood the scope of Part 11 to be very broad. We believe that some of those broad interpretations could lead to unnecessary controls and costs and could discourage innovation and technological advances without providing added benefit to the public health. As a result, we want to clarify that the agency intends to interpret the scope of Part 11 narrowly.”3
According to narrow scope as described in the US FDA “Guidance for Industry. Part 11, Electronic Signature —Scope and Application,” Part 11 applies only to records required by predicate rules. Computerized system life cycle records and IT process records are not required by predicate rules and are out of scope. These may be contrasted with records that are required by predicate rules, including batch, calibration, and laboratory product test records, and process validation and analytical validation data. The US FDA recommends that regulated companies determine, based on the predicate rules, whether specific records are Part 11 records and recommends documenting such decisions.3 Establishing processes to achieve this (e.g., defined in an standard operating procedure) is suggested.
Computerized systems supporting the supply of medicinal products to multiple markets must also comply with GxP regulations applicable in those countries and not only US FDA record requirements. The GAMP approach to computerized systems compliance and record and data integrity is designed to satisfy a broad range of international requirements.
Risk-Based Approach
Regulatory guidance from the UK Medicines and Healthcare Products Regulatory Agency (MHRA) and the US FDA are clear that formality and extent of controls for any records and signatures should be based on risk. The focus should be on patient safety, not on compliance. The approach described in this article is aligned with such risk-based thinking.
MHRA
“Controls should be proportionate to the risk considering the type of document and the methods used for distribution and approval… Aspects to consider when assessing risk include … whether there is a legislative requirement or GxP guidance for a signature. If there is, then the signature should be considered more critical and have proportionately greater control—for example when a QP [qualified person] certifies a batch of finished product to enable release for sale.”4
The GAMP approach to computerized systems compliance and record and data integrity is designed to satisfy a broad range of international requirements.
FDA
“We suggest that your decision on how to maintain records be based on predicate rule requirements and that you base your decision on a justified and documented risk assessment and a determination of the value of the records over time.”3
Critical Thinking
Inappropriate application of rigid GxP record approaches and rules designed for other situations and contexts, without critical thinking, can discourage or prevent the use of best practice techniques and tools with no concomitant benefit to product quality or patient safety.
There have been cases of the use of standard tools being disallowed, being used in ineffective ways, or being inappropriately customized to meet spurious and unnecessary expectations. This can lead to unnecessary costs, lower quality product, decreased flexibility, and inferior process control. It can discourage innovation and technological advance, and it may prevent IT, software, and quality professionals from selecting and using the most appropriate and effective tools and systems to perform their duties.
For example, it is incorrect and unhelpful to argue that Part 11 requires a data audit trail to be applied to changes to software code, maintenance of system configuration records, or maintenance of user access privileges. A data audit trail is simply not an effective mechanism in those cases. It is also incorrect and unhelpful to insist on unnecessary Part 11 signatures, where there is no requirement for regulatory approval or legally binding equivalent of the individual’s handwritten signature.
Other examples include printing maintained records in order to apply handwritten signatures, duplicating electronic records in a paper format, or duplicating other actions and activities in unnecessary and cumbersome paper processes.
System life cycle management and IT process management tools have been routinely used in other industries (including automotive, consumer electronics, finance, energy, military, and aerospace) for decades, but they are often underutilized or misused in the pharmaceutical and other life science industries due to conservative interpretations of regulations, a misguided perception of regulatory inflexibility, and a lack of understanding of modern software and quality assurance practices. A modern approach to the use of such tools is described in Appendix D9 – Software Tools of GAMP® 5 (Second Edition).1
Practical Implications for Quality, Cost, and Time to Market
This is not just an academic or theoretical discussion: It has important practical implications. Inappropriate requirements, rules, and practices that do not enhance product quality and patient safety may act counter to current good practice. This can lead to activities that are unnecessarily costly, time-consuming, or ineffective. These may also be potentially harmful by adding complexity, duplication of information, and unnecessary customization.
The US FDA has described how an excessive focus on compliance may divert resources and management attention away from investments in quality and toward compliance activities like documentation, which do not directly lead to improved quality outcomes.5 The US FDA has observed that a compliance-centric approach has not only hampered innovation in manufacturing and product development practices, it has also resulted in quality issues, and the perceived regulatory burden has contributed to outdated compliance practices.2
This is, therefore, not purely a matter of interpretation of regulation but selecting and applying methods and techniques that best serve the interests of the patient and the public. From the perspective of the public, patient, shareholders, colleagues, and other company stakeholders, we must apply critical thinking to make pragmatic and logical decisions. For example, a practical question to ask when applying critical thinking is: “What approach provides the most value and gives the best outcome for the patient, the shareholders, and other company stakeholders in general?”
It is more beneficial to apply modern processes using state-of-the art tools and automation (designed specifically to support IT and software activity) that deliver systems that work instead of costly, inefficient, and unnecessary practices. These practices may have a negative impact on quality and may be based on a dogmatic reading or misunderstanding of regulations and historical perception.
Such judgements are not purely academic; there is a key ethical dimension. Decisions we make as an industry can impact time to market, limit the availability of medicines to patients, and drive up the price of medicines unnecessarily. We must apply ethical critical thinking and choose the option that gives the greatest benefit to the patient and the public.
Benefits of Using Appropriate and Effective Tools
The approach described in this article supports effective system life cycle processes, including version, change, and configuration management; requirements management and traceability; testing; verification; and defect prevention.
Practical examples of the advantage of using standard and widely used tools include effective configuration management and data management, automatic version control with auditing capabilities, change management and documentation, automated testing, and continuous integration and efficient issue tracking. Tools support and ensure both accountability and traceability in the coding, testing, and deployment processes.
Code modification, managed through a robust commit process, is recorded with details of the who, when, and how of the modification. Automated checks are performed at each commit. This allows for immediate notification to the developer in case of errors or defects, supporting the identification and addressing of issues during the life cycle. Regression tests are built in and are routinely run. The transparency and accountability that such tools automatically provide greatly contribute to the overall quality control process.
Key operational processes—including incident, problem, and configuration management; security; performance; capacity; and cybersecurity threat management—can only be performed effectively by using the appropriate tools. Traditional paper-based approaches cannot provide the desired controlled state.
As noted by the Cloud Security Alliance (CSA), a leading international organization dedicated to defining and raising awareness of best practices for a secure cloud computing environment, issues impacting good software development practices that hamper secure and effective deployment are manual and haphazard coding, testing, deployment, and patching practices:
“Without automated quality checks, manual coding can easily result in poor performing and insecure software that needs rework. In addition, manual and poorly-timed testing reduces the chance that vulnerabilities will be identified before deployment. Manual deployment and patching practices can result in insecure software from being released to production.
“Automated security practices are the core of process efficiency because they can reduce manual processes, increasing efficiency and reducing re-work. Software quality can be bettered by improving the thoroughness, timeliness and frequency of testing/feedback. Processes that can be automated should be automated, and those that can’t should be automated as much as possible or be considered for elimination.”6
Conclusion
GAMP guidance makes a valuable distinction between GxP records supporting the medical product life cycle and required by a predicate rule, and non-GxP information, data, and artifacts that support computerized system life cycles and IT processes.
The unnecessary application of some customs and practices traditionally associated with GxP records, without critical thinking, to system life cycle and IT process records can lead to unnecessary costs and lower quality and process control. It can also discourage innovation, and may prevent IT, software, and quality professionals from selecting and using the most appropriate and effective tools and methods to perform their duties.
Patient safety, product quality, and data integrity are best achieved by managing computerized system life cycle and IT process records through use of current good IT practice and software engineering principles supported by effective standard tools.
About the Authors






