Global Collaborative Review: Understanding Overall Control Strategy and Patient Benefit-Risk

The pharmaceutical industry faces considerable challenges throughout the development, manufacturing, and supply of medicines, largely due to the intricate and divergent global regulatory landscape. The adoption of structured data standards and utilization of cloud-based platforms offer immense potential to overcome these challenges by facilitating faster and more efficient global collaborations between health authorities and industry. This potentially can be achieved by using a visual roadmap of the overall control strategy to help understand patient benefit-risk more effectively.
Worldwide, industry has many regulations to comply with and compliance to these regulations can be challenging.1 The process of preparing, submitting, and maintaining regulatory documents for product approval is a lengthy and expensive endeavor.
For many organizations, it is currently a manual process due to limited utilization of software tools to assist with these processes.2 Another challenge that industry faces is managing postapproval changes. This is a vital and complicated part of pharmaceutical manufacturing and regulatory compliance, which requires a thorough understanding of regulatory requirements, effective risk management, and careful planning.
These changes may take years before they are approved by regulatory agencies.1 In addition, the pharmaceutical industry must comply with inspections of manufacturing facilities by regulatory agencies to ensure alignment with GMP standards. The challenge is that preparing for inspections can be resource intensive, especially when multiple regulatory bodies conduct inspections at different times.
However, the burden of inspections can potentially be minimized by creating a standardized structure for the presentation of the marketing application dossier content. This standardized structure facilitates easier dossier navigation, helping the inspector quickly find the information and link to the underlying supporting data.
Another complicating factor arises from the varying rates at which new International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) members adopt ICH guidelines, as well as the interpretation of those guidelines by regulators from different countries. Additionally, there is a significant gap in the level of regulatory maturity from an innovation standpoint.
The complexity arises when reviewing innovative products, as there may be challenges in understanding the scientific principles behind the innovation, as well as the legal and regulatory requirements associated with them. These complexities can lead to differing perspectives on assessing risk, ultimately resulting in country-specific control strategies for new technologies.
A recent study conducted by the International Consortium for Innovation and Quality in Pharmaceutical Development (IQ) Control Strategy Global Harmonization Working Group examined 112 marketing applications encompassing both new synthetic and biological products from 11 companies.3 The study aimed to evaluate industry’s experience with divergences, raise awareness of common trends, and shed light on the implications for multiple stakeholders, including regulators, the industry, and patients.
Using agreed-upon criteria for core document acceptance, the average core document acceptance rate across the US, European Union (EU), Canada, and Japan is 54% for synthetic and biologic products. When this overall acceptance rate (54%) is translated into the likelihood of core documents being accepted by all four countries, it results in a probability of 8.7%. This underscores the subjectivity involved in determining an acceptable control strategy. A fundamental issue for control strategy divergence is that the current version of ICH M4Q(R1) does not explicitly state where or how an overall, end-to-end control strategy is described in a regulatory submission.4
There have been discussions about structured data and cloud-based submissions to streamline the regulatory review process. However, as data can be interpreted in different ways, it is important to balance data and narrative to understand the overall benefits and risks of the product to the patient. Thus, as part of this article, we explore each of these interconnected processes and present an innovative visual roadmap for the overall control strategy. This strategy uses structured data- and science-driven risk-based assessments to describe the product quality for the intended patient population within a cloud-based submission.
This concept of presenting structured data within the context of a narrative can serve as a valuable tool for facilitating collaborative reviews between sponsors and regulators. By integrating structured data with a coherent narrative, it can enhance the effectiveness of communication within the application process. This approach not only helps ensure clarity and understanding but also fosters a potential streamlined and productive exchange of information between the involved stakeholders. This contributes to a more efficient and informed decision of the application.
The Evolving Chemistry, Manufacturing, and Controls (CMC) Landscape
New modalities and innovative manufacturing approaches are driving a wave of innovation as biopharmaceutical manufacturers must adopt novel approaches that incorporate advancements in technology and new ways of working. For example, advanced therapy medicinal products (ATMPs), which include cell and gene therapies, and tissue-engineered products offer potential to treat diseases and conditions that were previously considered incurable or that have limited treatment options.
Other examples are advanced technologies that include continuous manufacturing, 3D printing, use of digital twins, and digital tools to enhance patient compliance for treatments. In addition, the use of AI and machine learning (ML) is now prevalent. These technologies are helping reshape drug discovery, development, manufacturing and analytics. They help evaluate chemical and biological data sets to predict drug candidates, identify novel drug targets, optimize drug formulations ensuring the stability and bioavailability of the product, and enable real-time monitoring and predictive maintenance of the manufacturing process.5
The combination of these advancements is transforming the way drugs are discovered, developed, manufactured, and delivered to patients. As advances continues in the pharmaceutical industry, it is a struggle for global regulations to adapt to these new technologies in a timely manner, which can cause delays to the introduction and supply of critical medicines to patients. However, there are several ongoing initiatives that seek to address these challenges. For example, the revision of ICH M4Q (R1) was endorsed by the ICH management committee and the concept paper was published 15 November 2021 (ICH M4Q(R2)).6
The concept paper for ICH M4Q(R2) has several promising topics, which include improving registration and life cycle management, leveraging digital technologies, and accelerating access of medicines to patients. Also encouraging are global convergence of science- and risk-based approaches, enabling efficient use of digitals tools and benefit–risk considerations. One of the most significant topics is the proposed overall control strategy, which the concept paper states “should be the backbone of the revised M4Q structure.”6
Structure Product Quality Submissions
In a press release on 3 June 2020, the ICH Management Committee introduced a new topic called Structured Product Quality Submissions.7 The use of structured CMC data for regulatory submissions will be a paradigm shift in how industry submits CMC information to regulators.8 Several data standards will impact the CMC domain.
ISO IDMP
One example is International Organization for Standardization (ISO) Identification of Medicinal Products (IDMP), which is designed to establish a common framework for the identification, documentation, and exchange of information on the identification of drug products.9
The goal is to improve the accuracy, consistency, and interoperability of data related to pharmaceutical products throughout their life cycle by structuring and standardizing product information.10 Although IDMP is not exclusive to the quality aspects of a regulatory application, the standard includes a significant number of CMC-relevant attributes, including details about the active ingredient(s), strength, dosage form, route of administration, and packaging.
IDMP promotes global harmonization of product information standards, making it easier for pharmaceutical companies to submit consistent data to multiple regulatory agencies worldwide. Implementation of ISO IDMP standards is an ongoing process. Regulatory agencies worldwide are gradually adopting these standards and pharmaceutical companies are working to align their internal data management systems with IDMP requirements.
PQ/CMC
Another data standard being discussed is the US Food and Drug Administration (FDA) Pharmaceutical Quality - Chemistry, Manufacturing & Controls (PQ/CMC). It was introduced in the Federal Register in 2017 to present structured data elements and controlled terminology for Module 3.11 PQ/CMC uses the established healthcare data exchange standard from Health Level 7 (HL7), called Fast Health Interoperability Resources, which is widely in use in the greater healthcare ecosystem but is being newly adopted for pharmaceutical and regulatory applications.12
The overall goal of the PQ/CMC project is to support application review by standardizing language and terminology to allow for preprocessing of data, population of review templates, and aggregation of data for inspections and general data retrieval.13
PQ/CMC was designed and developed to be implemented in support of product assessment. Therefore, it provides detailed data element definitions, supporting controlled terminology, and the mandatory/optional aspect for each element to allow for consistent and standardized data in various sections of Module 3. The FDA attempted to map PQ/CMC data elements to the ISO IDMP standards, which showed similarities and differences between the two initiatives.14
Need for Data Standards Alignment
Although there is some overlap in both concepts, the relationship between these emerging data standards and the work that will be pursued under the ICH Structured Product Quality Submissions guidelines is unclear at this time. The maximum benefits of these efforts can be achieved by ensuring alignment and agreement on one data standard, which can reduce divergence across the regions. Notably, the use of structured data enables CMC submissions to be amenable for cloud-based technologies.
Cloud-Based Platforms
The introduction of cloud-based, data exchange platforms into the pharmaceutical landscape have emerged as a transformative tool. For example, the nonprofit industry association Accumulus Synergy is developing a cloud-based platform to enhance data information exchange for submission content and collaboration for regulatory submission pathways.15 The adoption of cloud-based platforms within the pharmaceutical sector has many advantages, especially when it comes to fostering collaboration across diverse teams, organizations, and geographical boundaries. This accessibility transcends physical and geographical constraints, enabling real-time global collaboration in a protected shared environment (see Figure 1).
Currently, industry has collaborative initiatives such as Project Orbis,16 the Access Consortium,17 and the International Coalition of Medicines Regulatory Authorities (ICMRA).18 One of ICMRA’s active working groups is Pharmaceutical Quality Knowledge Management (PQ KMS). This group has conducted postapproval collaborative pilots; engaged and coordinated with external organizations such as ICH, International Pharmaceutical Regulators Programme (IPRP), and Pharmaceutical Inspection Co-operation Scheme (PIC/S); and developed high-level reflection papers to promote collaboration.19 These initiatives provide an excellent starting point for global harmonization efforts across the regulatory agencies and are currently accomplished without the use of cloud computing technology.
The use of a cloud-based platform enables further collaboration and facilitates a greater number of regulators to participate in these initiatives in a seamless manner. However, there are challenges to adopting cloud-based technologies, such as cybersecurity concerns and country-specific legislations. For example, the EU’s data privacy and security law, General Data Protection Regulations (GDPR), is one of the toughest privacy and security laws in the world20 GDPR requires that any information collected from citizens of the EU must reside in servers located in EU jurisdictions or in countries with a similar scope and rigor with their privacy laws.
Automated Data Interpretation and Computer-Aided Data Analysis
As we approach this exciting stage for industry, there should be caution around the interpretation of data using automated tools. Data in its raw form can be interpreted in a myriad of ways, but to fully grasp its scope and utility, context is needed. For sponsors, context is needed to distinguish nuances in the data that are relevant to the underlying production approach and technology. Additionally, the degree to which evidence can be supplemented by prior knowledge could drive significant differences in the data package between sponsors, which would be imperceptible to the reviewer with significant context in the application.
For health authorities, the incorporation of structured data into CMC submissions holds the potential to leverage data analytics and AI/ML to enhance the efficiency of regulatory reviews. A case in point is the FDA’s adoption of the Knowledge-Aided Assessment and Structured Application (KASA). KASA establishes rules and algorithms for risk assessment, control, and communication related to product, manufacturing, and facilities.21 KASA also facilitates computer-aided analysis of the applicant’s submission to FDA’s institutional knowledge instead of relying solely on the summarized narratives provided in submissions.
Balance of Data and Narrative
Although the comparison of dosage forms, manufacturing processes, and other relevant CMC attributes can provide valuable data-driven assessments, CMC attributes should be considered within the context of the intended patient population to ensure that a pharmaceutical product is safe and effective. For example, patient-centric factors such as disease state, treatment duration, age, and characteristics of the patient population are important to understand the overall risk of the administered drug product.
It is therefore critical to have a balance between data and narrative to understand the benefits and risks of the product to the patient. One strategy is to use the overall control strategy to bridge data and narrative to demonstrate the overall benefit–risk details of the medicine to the patient population.
Overall Control Strategy
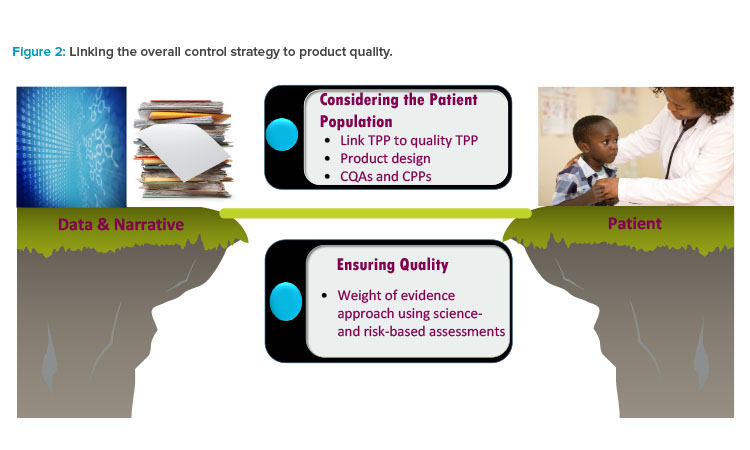
The understanding of the overall control strategy plays a pivotal role in the product’s risk assessment and in safeguarding drug product quality (see Figure 2). It also ensures that the quality attributes and performance characteristics of the product are consistently maintained throughout its entire life cycle.
The concept and aspects of the control strategy are addressed in several ICH guidelines, including ICH Q8–Q14 and in the concept paper for ICH M4Q(R2).22, 23, 24, 25, 26, 27, 28 The ICH guidelines emphasize the importance of designing a robust control strategy as part of the pharmaceutical development process. This ensures that pharmaceutical companies have a comprehensive plan in place to consistently deliver safe and effective medicines to patients. At the same time, it allows them to consider scientific understanding, risk assessment, and regulatory compliance.
The control strategy is a pivotal component of pharmaceutical development, representing a structured approach to ensuring product quality, safety, and efficacy throughout its life cycle. It is fundamentally linked to the concept of quality by design (QbD), which promotes a proactive and science- and risk-based approach to development strategies for the CMC controls of the product.
The sponsor’s institutional knowledge is combined with specific drug product information, including:
- The target product profile (TPP), which identifies the characteristics of the drug product necessary for the patient population and disease indication
- The patient quality target product profile, which outlines the quality characteristics of the drug product necessary to meet the TPP
- Critical quality attributes (CQAs)
- Critical process parameters (CPPs)
These specifications, as well as continual improvement, are key elements of the control strategy. Its primary objective is to ensure that the drug product consistently meets its intended quality attributes, thus delivering safe and effective medicines to patients worldwide.
| Risk Assessment | Risk Control | Result of Quality Risk Management (QRM) Process/Risk Review | |||
|---|---|---|---|---|---|
| Hazard Identification | Risk Analysis | Risk Evaluation | Risk Reduction | Risk Acceptance | Risk Rating |
| Risk Rating | Risk Definition |
|---|---|
| Low | Low impact on quality affecting safety and efficacy of medicinal product |
| Medium | Medium impact on quality affecting safety and efficacy of medicinal product, if no further controls are put in place |
| High | High impact on quality affecting safety and efficacy of medicinal product, if no further significant/novel controls are put in place |
Overall Control Strategy with the Use of Structured Data
One innovative approach to understand product quality for the intended patient population is by creating a visual roadmap of the overall control strategy. The roadmap is created using structured data and risk-based assessments for cloud-based submissions. This entails organizing and presenting data in a structured and interconnected manner that provides reviewers with a holistic overview and in-depth understanding of the overall control strategy.
Tables 1 and 2 illustrate an example of how risk can be communicated to regulators by using quality risk management (QRM) principles from ICH Q9(R1).23 The evaluation of the risk to quality should be based on scientific knowledge and ultimately linked to the protection of the patient.
In the example shown in Table 1, the sponsor can communicate risks qualitatively by initiating a QRM process that describes the risk assessment, risk control, rating output, and review for the product. Table 1 provides an opportunity to discuss data and narratives that support a science-driven, risk-based approach that describes the risk in terms of how it can impact quality of the product. Table 2 provides an example of risk definitions that can be used for risk communication.
The goal for this approach is that risks are systematically and comprehensively evaluated by a weight of evidence approach to the sponsor’s prod-uct-specific knowledge, overall control strategy, and benefit–risk assessment of the product with the intended patient population.
For example, risk should not be evaluated in isolation, but understood and assessed against the overall control strategy. This should be done with the understanding of how that risk impacts quality in terms of safety and efficacy for the intended patient population. This approach brings transparency and clarity to the dossier, enabling regulators to navigate through the application of the pharmaceutical products more efficiently and understand the risks in a patient-centric manner. It potentially can be a communication tool between regulatory bodies and the industry, fostering a collaborative environment for review and understanding of product quality.
Although the ICH guidelines stress the importance of developing a robust control strategy as part of the pharmaceutical development process, there is no current regulatory guidance that clearly states where or how a comprehensive end-to-end control strategy should be described in a regulatory submission. One innovative approach to understand product quality for the intended patient population is by creating a visual roadmap of the overall control strategy, using structured data, and providing a risk assessment for cloud-based submissions.
This concept has been initiated by the ISPE Risk-Based Quality Overall Summary (QOS) Working Group. The approach entails organizing and presenting data in a structured and interconnected manner that provides reviewers with a holistic overview and in-depth understanding of the overall control strategy.
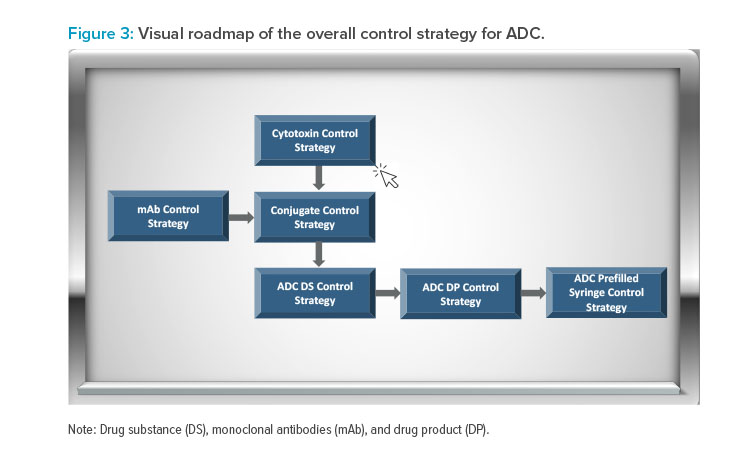
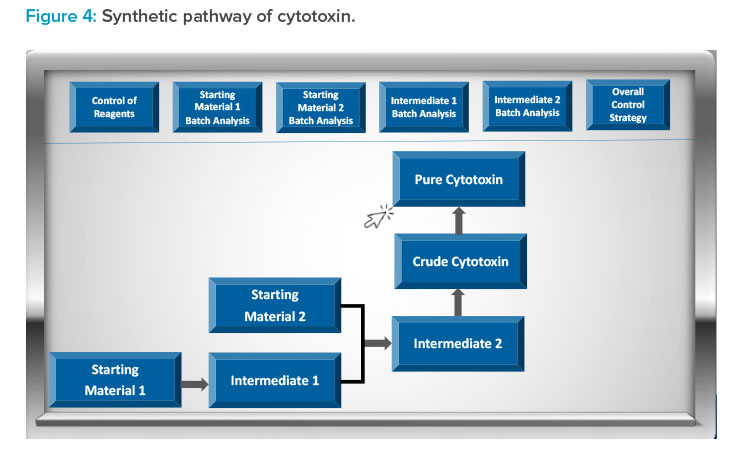
Figure 3 presents an example of the visual roadmap concept for the overall control strategy of an antibody–drug conjugate (ADC). In a cloud-based environment, reviewers have the flexibility to choose specific sections of the control strategy for review. For instance, as depicted in Figure 3, the re-viewer can opt to examine the cytotoxin control strategy by selecting the box. This will provide further details of the cytotoxin synthetic pathway and controls, as shown in Figure 4. If the reviewer wishes to delve deeper into the pure cytotoxin, they can access additional information, which is presented in Figure 5.
This interface empowers the reviewer to explore various options, gaining a comprehensive understanding of the cytotoxin’s quality. Reviewers can also download data for independent assessments while having a clear grasp of the innovator’s provided control strategy. This interactive approach allows for a systematic exploration of the application. This facilitates an in-depth evaluation of the product’s benefit–risk profile for the patient population. It’s important to note that the ADC example illustrated in Figures 3–5 serves as a conceptual representation.
The ISPE Risk-Based QOS Working Group plans on illustrating this concept in a cloud environment by structuring a mock submission. This demonstration within a cloud environment shows one example of the potential use of structured data in cloud-based submissions.
Conclusion
The pharmaceutical industry plays an indispensable role in ensuring patients worldwide have access to essential medicines. However, it faces considerable challenges throughout the development, manufacturing, and supply of medicines. This is largely due to the intricate and divergent global regulatory landscape. The adoption of structured data standards and utilization of cloud-based platforms offer immense potential to overcome these challenges by facilitating faster and more efficient global collaborations between
health authorities and the pharmaceutical industry. It is crucial to strike a balance between data-driven assessments and the narrative context to comprehensively evaluate the benefit–risk profile of the medicine for the patient population.
One innovative concept is the creation of a visual roadmap for the overall control strategy using structured data and science- and risk-based assessments that describe product quality for the intended patient population in cloud-based submissions. This approach allows reviewers to gain a holistic understanding of the control strategy. It also allows each piece of information linked to describe the medicine’s quality in terms of benefit and risk for the specified patient population. Ultimately, these improvements may translate into better healthcare outcomes for patients across the globe, reaffirming the industry’s crucial role in delivering quality medicines to patients.

Acknowledgements
The authors thank Forian Lengyel and Dhiraj Behl for their support with manuscript preparation and review.


