
Considerations for a Decentralized Manufacturing Paradigm

The biopharmaceutical industry must develop and implement innovative ways of working to be effective and efficient in the current healthcare ecosystem, in which high-quality medicines, adaptability, and assurance of supply are of critical importance. There are regulatory strategies and technologies emerging to address these challenges, but further progress must be made to fully harness the advantages of advanced and decentralized manufacturing techniques.
In comparison to traditional large-scale manufacturing operations, decentralized modes of manufacturing can be more agile, more transportable, and tailored for specific modalities or production processes. However, although the supporting technologies needed to enable decentralized manufacturing have continued to advance over the past decade, the regulatory expectations for submission data packages must also evolve, using the technology transfer of a biologic as a relevant example, to make implementing decentralized manufacturing more practical and attainable for manufacturers.
This article presents relevant insights on the current regulatory and technical landscape for decentralized manufacturing, with select examples of current applications, and discusses perspectives on evolving and adapting the current regulations to meet future capabilities.
Advanced Manufacturing
In the biopharmaceutical industry, “advanced manufacturing” refers to the operating models and supporting technologies that aim to transform or modernize the production of therapeutic products.1, 2, 3 Though traditional large-scale batch modes of operation can be used for many small and large molecule drugs, such that manufacturing strategies can be designed as fit for purpose, alternative strategies to traditional manufacturing methodologies are increasingly needed to operate more efficiently, keep pace with market demands, and enable the commercialization of advanced or personalized therapies. Specifically, in biopharmaceutical development, there is a continued trend of new modalities with increasing complexity and smaller batch sizes, which requires retooling traditional manufacturing approaches for those applications.
The COVID-19 pandemic highlighted the need for responsive and agile manufacturing systems that can reliably perform and meet supply chain demands in a rapidly changing environment. In response, modes of manufacturing that are decentralized, transportable, and increasingly configurable have emerged as areas of interest and investment for both industry and regulators. These new operating models can help enable and accelerate the efforts of Pharma 4.0™ toward the implementation of smart manufacturing, digitization, and automation.4, 5 Additionally, reducing shipping limitations—such as packaging, storage, and transportation durations—helps speed up patient access in certain instances while ensuring there is no negative impact to the product’s critical quality attributes (CQAs).
Although the novelty of these modes of manufacturing has led to some definitional ambiguities, initial working definitions and terminology have been drafted by several regulators, trade organizations, and subject matter experts. There are several operating models that can potentially be categorized under “decentralized” manufacturing, including distributed manufacturing (DM), modular manufacturing (MM), and point-of-care (POC) manufacturing. The following working definitions are useful in establishing foundational understanding; however, harmonization of terminology is ultimately needed to develop cohesive and supportive regulatory frameworks across regions.
Herein, DM refers to a manufacturing model in which many units or facilities are disseminated across different geographical areas6, 7 DM facilities may produce similar or identical products under a unified pharmaceutical quality system (PQS) or operate as independent units with different outputs.6 MM describes a platform in which unit operations and/or manufacturing activities are conducted in interchangeable components or modules.8, 9 Modules can be switched in or out to meet the current needs of the facility, including single-use materials as appropriate, to complete different types of production.
Currently, most modular facilities operate in a single fixed location that leverages the production flexibility granted by the modules, but modular operations may also be transportable and capable of enabling DM in a portable-on-demand (POD) format.10 Similarly, POC manufacturing describes production at or near the location of the patient. POC manufacturing solutions can consist of modular and distributed approaches in which modules or units are geographically dispersed across local patient hubs.
Fit for use is a critical factor that companies must evaluate when selecting a manufacturing operating model. In particular, DM and POC manufacturing models are considered a natural fit to produce many cell and gene therapies. As a result, there has been substantial prior discussion on DM and POC manufacturing that has focused on solving for logistical challenges in producing highly individualized therapeutics. However, DM, MM, and POC manufacturing may be suitable for other types of biologic products as well, including those that are currently produced in large-scale, centralized manufacturing facilities, such as monoclonal antibodies (mAbs).
For example, fill-finish operations for products using prefilled syringes can be performed close to the point of distribution or point of care to address transport challenges caused by stability issues that require ultra-low-temperature storage and shipping conditions. Similarly, having multiple sites of manufacture or distribution, or flexible modules that enable reconfiguration of a facility, can help ensure patient supply close to the point of use and can manage localized drug supply shortages across product types.
Here we focus on the potential uses for DM, MM, and POC for biologics, highlighting emerging technologies, proposed changes to the regulatory framework to enable implementation, and anticipated future developments in the technological and regulatory landscapes.
Emerging Technologies and Recent Applications
Agility, flexibility, and reproducibility of a biomanufacturing operating model are enabled and empowered by a variety of advanced technologies including, but not limited to, single-use technology (SUT), process analytical technology (PAT), continuous manufacturing (CM), POD, and Pharma 4.0™ technologies. These emerging technologies promote local and highly flexible production that is as robust as, or in some cases superior to, traditional testing and manufacturing in terms of consistency and control through a number of mechanisms. However, in addition to the advantages of the emerging technologies, there are also various challenges for industry and regulators when incorporating these newer concepts into the traditional regulatory submission framework during the product life cycle.
An overview of the concepts, advantages, technical challenges, and considerations associated with the implementation of the innovative technologies is provided next. Select applications of the aforementioned technologies in the context of decentralized manufacturing are provided in Table 1. Previous articles have discussed these applications and biopharmaceutical industry examples at greater length and detail.3 However, herein, we summarize prominent enabling technologies; offer some recent examples of real-world applications in POC, MM, and DM complementary to technology transfers; and provide advantages and challenges to implementation in the current manufacturing, technological, and regulatory environments.
| Manufacturing Type | Company/Institution | Product Information | Key Facts of Facility and Manufacture | Location | Phase |
|---|---|---|---|---|---|
| Point-of-care manufacturing (POC) | University of Maryland, Baltimore County33 | Granulocyte colony-stimulating factor (G-CSF) |
|
Can be installed in all local hospitals, fire stations, pharmacies, and other potential evacuation centers | Development |
| POC | Dmitry Rogachev Center and University Hospitals Seidman Cancer Center34 | Anti-CD19 CAR (CAR19)-T cells |
|
Moscow, Russia, and Ohio, US | Phase 1 |
| POC | CellPoint B.V.35 | Cell therapy (GLPG5102) |
|
Europe (multicenter study) | Phases 1 and 2 |
| POC | Orgenesis36, 37 | Cell therapy |
|
Regional hub supply centers in the EU and US that will serve medical centers within a four-hour travel radius | Development |
| Modular manufacturing (MM) | ADMA Biologics, Wildlife Pharmaceuticals, Emergent BioSolutions, BPI Labs, Singota Solutions, WuXi Biologics, FUJIFILM Diosynth Biotechnologies, and Genentech38 | Across dosage formats and sizes including biologics |
|
US, South Africa, Canada, and China | Clinical/ Commercial |
| MM | Janssen Pharmaceuticals in collaboration with Legend Biotech39 | Cell therapy (CAR-T cells) |
|
New Jersey, US | Clinical |
| MM | Sanofi40,41 | Vaccines |
|
Neuville-sur- Saône, France | Intended commercial |
| MM | Amgen42 | Biologics (multiple products) |
|
Singapore | Commercial |
| MM | Genentech43 | Biologics (multiple products) |
|
California, US | Intended commercial |
| MM | Eli Lily and Company 44, 45 | Synthetic drug substance |
|
County Cork Ireland | Development |
| MM | Pfizer46 | Sterile injectables |
|
Michigan, US | Intended commercial |
| MM with distributed manufacturing (DM) | BioNTech47, 48 | Vaccines such as Comirnaty |
|
Rwanda, Senegal, South Africa, and Australia | Intended clinical/ commercial |
Enabling Technologies for MM and POC Manufacturing
Continuous Manufacturing
Description
CM is a single integrated process in which the input materials are continuously added and transformed during the process, with continuous output. CM can be applied to an individual unit operation; however, it most often refers to an integrated process consisting of two or more unit operations. CM is applicable to drug substances and drug products for synthetic entities and biologics.11, 12, 13, 14, 15, 16, 17
Advantages and Considerations
CM improves manufacturing efficiency and flexibility. It reduces the manufacturing footprint, provides “right-size” production scales by adjusting process run times to meet demand, expedites technology transfers due to similarities in equipment and scale across sites, and is amenable to PAT, which supports process validation and continued process verification (CPV). However, it may present increased system and validation complexity, e.g., PAT validation, and any upfront investment in equipment and expertise needs a business justification. Further, because CM is newly emerging, there is a need for more highly trained staff. It also may not be available at all contract manufacturing organizations (CMOs), especially for biologic drug substances; thus, outsourcing may not be an option.
Advanced Aseptic Technologies
Description
An advanced aseptic technology is an aseptic process or system in which design and automation are used so that direct human intervention from operators is not required or permitted during processing. Examples include a closed aseptic filling system, isolator, and robotic arm for aseptic filling.18, 19, 20, 21, 22, 23, 24, 25, 26
Advantages
In advanced aseptic technologies, the equipment provides a robust sterile manufacturing environment and transportable solutions for DM aseptic manufacturing operations. These technologies also offer a combination of closed, connected process and the centralized control facilitates improved efficiency, safety, and compliance.
Challenges and additional considerations
In these technologies, minimizing microbial contamination relies on a foundation of system design and controls and requires careful planning, environmental monitoring, specialized facilities and equipment, and trained personnel. Integration, such as incorporating an isolator into a lyophilizer, can be complex and cycle time can be extensive. Closed restricted-access barrier systems (RABSs) and isolators are expensive compared to standalone systems. Additional stability studies may also be needed to ensure there is no impact to product quality from decontamination or cleaning products like hydrogen peroxide.
Portable-on-Demand Format
Description
POD refers to a type of “autonomous and portable” manufacturing facility with one or multiple units that house a defined set of pharmaceutical operations and that is variable in size. Examples include platforms as small as autonomous units placed within a facility and mobile production trailers for compounding to platforms as large as a prefabricated, self-contained GMP facility.3,27
Advantages
POD increases manufacturing flexibility, speed, and consistency. Duplication enables rapid scale-up and scale-out, new facilities can be constructed in less than 12 months, and isolated operation avoids cross-contamination. POD is suitable for a wide range of products, including oral solid dosage forms, mAbs, and cell therapy. The fit-for-purpose and reduced-size POD units promote green manufacturing. It can add functionality by establishing a new facility or repurposing or reconfiguring an existing facility.
Challenges and additional considerations
Challenges exist in understanding and mitigating mobility-related quality risk factors and adjusting PQSs to include additional engineering control considerations. POD creates new challenges in meeting existing regulatory standards such as the registration of a physical address. Due to the limitation of instrumentation and size when analyzing a sample on-site, analytical technologies supporting POD require further development and are continuously evolving. Regulatory divergence and local expectations may also serve to limit the extent to which the benefits of “lift and shift” or “duplicate and move” can be realized.
Single-Use Technology, System, Assembly, or Equipment
Description
SUT or SUSs are most commonly constructed with polymeric components, creating a system or unit operation that is designed for one-time or a sin-gle-campaign use and is subsequently discarded. Examples include bioprocessing bags (replacing glass flasks and tanks), single-use bioreactors and accessories (replacing stainless steel bioreactors), aseptic connectors, and transferring assemblies and filters.12, 28, 29, 30, 31
Advantages
SUT creates closed systems, which separates operators and rooms, enables aseptic transfer between containers, and mitigates the risk of cross-contamination. It also reduces environmental impact because it requires fewer chemicals and less high-purity water and heat. SUT has transformed bioprocessing in terms of design, scale, operation, control, and speed of incorporating innovations. It shortens the time required to build a manufacturing facility and develop a manufacturing process. In addition, SUT reinforces the GMP emphasis on equipment qualification, reduces facility footprint, downtime, changes over time, and increases manufacturing capacity.
Challenges and additional considerations
With SUT, control strategies and supply assurance are heavily dependent on supplier collaboration, for example in the areas of quality systems, components and container qualification, change management, and supply chain management. Risk assessments require additional considerations such as chemical compatibility between process solution and materials of construction, risk of extractables and leachables, and establishing material interchangeability and second sourcing to enable uninterrupted supply.
A functional equivalency to traditional approaches may be challenging to establish with SUT because of its limitations in oxygen and heat transfer, or limited usability in high-temperature and high-pressure processes due to materials of construction or limitation in available sizes to accommodate the same processing volumes as traditional equipment. Single-use assemblies may pose risks or cause defects due to material manufacturing, assembling processes, or transportation. Further, traditional manufacturing may need to be in place to mitigate the impact of unavailability of SUSs due to quality or supply issues, and strategies are required for the treatment, handling, and disposal of post-use waste.
Industry 4.0 Technologies
Description
Industry 4.0 technologies deployed in the biopharmaceutical industry center on the concept of capturing and connecting all relevant data, and applying advanced analytics to the connected data to produce process insights so that ultimately the manufacturing plant can be automated with less human interaction. These technologies can include Internet of Things, artificial intelligence, robotics, and advanced computing.5, 13, 27, 32
Advantages
Industry 4.0 technologies enhance manufacturing efficiency, manufacturing flexibility, and product quality. PAT enables real-time monitoring, which reduces or eliminates conventional in-process testing, enables real-time release testing (RTRT), and reduces production cycle times. Large-scale data analytics support process validation and CPV. Digital twins can be applied to better understand, evaluate, predict, and optimize process performance.
With these technologies, there’s an abundance of data collected throughout the product life cycle, across sites, and across products, all with the potential to reduce certain repeated work in technology transfer. Feedback controls can be enabled through a combination of automation and predictive modeling. Compliance with quality and safety standards can be improved through the use of automation and robotics.
Challenges and additional considerations
However, Industry 4.0 technologies face a few challenges. Implementation can be challenging due to legacy systems integration, the disparate nature of hardware and software offered by a range of different vendors, enabling and/or establishing robust system foundation, and cultural change. They also potentially require a large initial capital investment and additional education and training to develop or upskill talent. Finally, analytical solutions, hardware, and software are still evolving to meet unprecedented levels of requirements in terms of functionality, robustness, validation, safety, and security.
Re-Envisioning Technology Transfer of a Biologic to New Manufacturing Sites
Within the biopharmaceutical industry, a philosophy held close by regulators and sponsors alike is that “quality should be built into the product, and testing alone cannot be relied on to ensure product quality.”49 However, regardless of the quality built into a product by enhanced understanding of product CQA and critical processes and the material parameters that impact those CQAs, a significant amount of data generated by manufacturing and testing must be submitted by sponsors to gain health authority approvals.50
The Past and Present
Historically, sponsors face the challenge of determining the amount of prior knowledge to be included in regulatory submissions, 52 whereas regulators are hesitant to acknowledge or place value upon submitted prior knowledge. Thus, sponsors may end up repeating studies that could be viewed as confirmatory, or even redundant, to enable chemistry, manufacturing, and controls (CMC) changes. This can result in the generation of new data for a specific monoclonal antibody (mAb), for example, when the sponsor already has existing data on similar platform products and processes that supports the fact that the CMC change will result in a product of comparable quality.
We will use technology transfer from an approved sending site to a proposed receiving site as an example. First, facility fit and gap assessments of the process between the sending site and receiving site must be conducted. Then substantial time and resources are needed to run the same manufacturing process at the receiving site and test the resulting product in quality control (QC) laboratories or use emerging technologies, such as PAT or RTRT, to generate release and characterization data demonstrating analytical comparability.
Subsequently, the new data is authored into Module 3 (Quality) of the Common Technical Document and submitted separately to multiple health authorities worldwide for approval. The sponsor then waits to distribute product with the change(s) implemented over an approval window of four months—as in the case of the US Food and Drug Administration (FDA)—to six years or more—as in the case of health authorities that require major market approvals prior to submission, and/or the submission of data from more post-change lots and stability data over longer durations of time.
The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Q12, Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management (2019), aims to simplify and harmonize life cycle management by identifying the types of changes that necessitate regulatory submission and by establishing risk-based reporting categories.53 However, endorsement, adoption, and implementation of ICH Q12 has been challenging for regulators and sponsors when it has been restricted by local legislation or differing health authority feedback on a single science-based and risk-based data set. Consequently, sponsors continue to face hurdles, such as variable regulatory review timelines and heterogeneous supporting data packages for postapproval change management.54
Although a complete package of fresh data is currently required for each mAb introduced to a new manufacturing site, it is unclear if this is a necessary or sustainable process for obtaining individual health authority approval for every technology transfer. Furthermore, this process becomes increasingly arduous in the context of DM, in which multiple manufacturing sites are generating data for the same or similar mAbs. Put simply, in situations where a sponsor has extensive manufacturing history, robust platform data, and a strong understanding of CQAs, the current regulatory framework affords no relief in Module 3 preparation by sponsors or review by health authorities. It takes the same amount of time for such a sponsor to get product to patients as it would for a sponsor who does not possess such process or product understanding.
This does not have to be the case because the foundation for science- and risk-based approaches exists. The ISPE Good Practice Guide: Technology Transfer (Third Edition) advocates for balance between risk management, resource management, and regulatory expectations.55 It also aligns with science- and risk-based quality by design (QbD) principles described by ICH and recognizes that knowledge management and a robust quality culture are critical to successful technology transfer.55
The Parenteral Drug Association (PDA) Technical Report No. 65: Technology Transfer (revised 2022), aims to standardize the approach to technology transfers, which would include conducting risk assessments, process comparisons, and knowledge transfers.56 ICH Q9(R1), Quality Risk Management, describes how quality risk management should be used in the impact evaluation of proposed CMC changes, and in determining the appropriate actions needed prior to implementation of changes, including testing, (re)qualification, (re)validation, or communication with regulators.56, 57
Practical utilization of these concepts to a larger extent within a more flexible regulatory framework could result in re-envisioned or nontraditional approaches to Module 3 preparation and review, enabling more efficient postapproval change management. This is particularly applicable in the setting of DM or POC manufacturing, for which the current regulatory framework places prohibitive hurdles to implementation. A science- and risk-based approach can and should be taken toward the data sets that need to be generated and submitted to health authorities to prove that quality is built into the product; specifically, that the biologic product manufactured from the new receiving site is analytically comparable to the product manufactured from the approved sending site.
Legal hurdles also exist, for example, in countries where health authorities prohibit dual sourcing, meaning the sourcing of a commercial product from more than one manufacturing site. Although it is acknowledged that the laws and regulations in many jurisdictions were not written for the potential of DM or POC manufacturing, such legal and regulatory hurdles are prohibitive of sponsors creating a DM or POC network, where similar or “sister” sites are used in different locations.
The Realized Potential
The COVID-19 pandemic highlighted the capability of sponsors and regulators to take science- and risk-based, nontraditional, approaches, which resulted in new perspectives. Both sponsors and regulators employed these approaches, which, in combination with close communication, expedited authoring and review durations, resulting in the accelerated use of COVID-19 vaccines manufactured by, for example, Pfizer and Moderna.58
As an additional example, to increase manufacturing network capacity for the manufacture of a third-party COVID-19 therapy, Amgen engaged the US FDA regarding a science- and risk-based strategy for a mAb drug substance technology transfer.59 Drug substance process validation data were generated in parallel to FDA review of meeting materials and a subsequent comparability protocol (or postapproval change management protocol [PACMP]). A final submission was negotiated to the downgraded reporting category of a change being effective in 30 days (CBE 30) supplement to submit the process validation data with reduced analytical comparability data.
A preapproval inspection (PAI) of the receiving site was not conducted by the FDA, likely due to the robust and credible historical clinical manufacturing experience of the mAb at, and the prior inspection history of, the receiving site.60 This strategy enabled the ability to distribute product to the US market with the change implemented eight months earlier than with traditional Module 3 content and reporting categories. Applying this type of flexible approach to postapproval change management for DM or MM, as well as for other appropriate site transfers, would ensure that the lessons learned from the pandemic are utilized effectively.
The Future
By applying the science- and risk-based strategies aforementioned, it is clear that more efficiencies can be gained, including lessening the review burden on health authorities. Although such strategies would directly enable sponsors to submit postapproval variations sooner, they would not immediately reduce Module 3 content. The success of science- and risk-based process, analytical, and regulatory strategies is contingent upon many important factors (the “how”).
To demonstrate to regulators that a process is understood and in control, CQAs of the product will not be negatively impacted, and the safety and efficacy of the product will be maintained in patients, the sponsor must provide to the regulators detailed process and analytical comparisons (pre-change compared to post change), clearly define the systems (i.e., the process, critical process parameters, raw materials, components, equipment, environment, testing methodologies, and/or technologies) from which the data sets are generated, and provide historical data (in the form of clinical, process development, process characterization data, and/or platform data) that support the change.
The concept of MM is a straightforward illustration—whatever is “modular” about the manufacturing, whether it be an entire room or a defined system within the manufacturing line, the module or system can be defined in narrative, images, and data. Then, this module or system, with defined inputs and environmental conditions under the PQS, can be used anywhere because the appropriate controls would be in place.
However, significant regulatory barriers surrounding this approach still exist, which may impede sponsors’ ability to implement. Notably, regulators require sponsors to identify all sites and site addresses as part of the application. If a site becomes mobile, either the definition of a site or reporting requirements for changing the location of a site will need revision.3. Furthermore, regulators may need to re-envision the requirements for and the conduct of inspections.
Sponsors would need to invest upfront in a submission that included extensive data supporting their change (such as a technology transfer) by defining and comparing processes and systems in detail, as well as referencing supportive data in the form of prior knowledge. In the future, if changes were compared and deemed the same (low risk) or similar (low to moderate risk), a reduced data package could be submitted, ideally using a reduced reporting category, and without needing to meet with a health authority or use a PACMP.
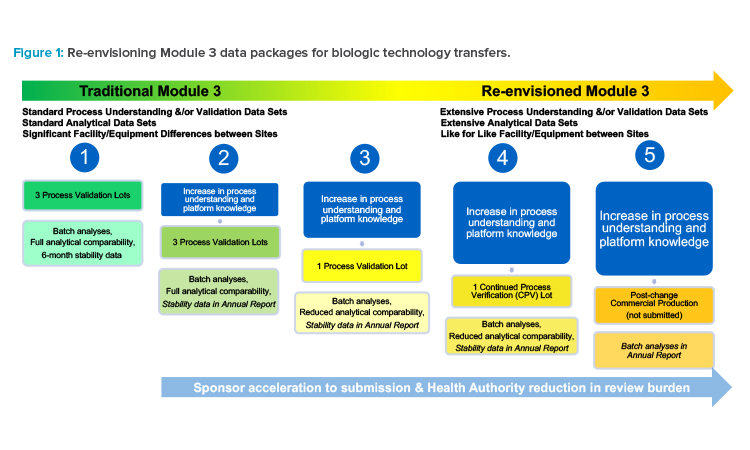
Such subsequent postapproval submissions would only include process validation data and reduced analytical comparability data, with a commitment to provide stability data in an annual report or notification (see scenario 3 in Figure 1). Or, instead of manufacturing and testing three commercial lots to support a traditional process validation at a new site, a single commercial lot could be manufactured with a medium- to low-risk change implemented, as part of CPV (see scenario 4 in Figure 1), demonstrating that even with the change implemented, the process remains in a state of control because quality is built into the product61 and the resulting product meets the quality target product profile (QTPP).
A site could initiate commercial production with the change implemented, and only batch analyses data could be submitted to health authorities after implementation in an annual report or notification. Of course, this would be contingent upon the comparisons made within that submission to the previously submitted processes, the capability (robustness) of the process, and justified applicability of the data sets (see scenario 5 in Figure 1).
The Impetus
Drug shortages continue to pose a challenge across the biopharmaceutical industry, resulting from delayed or discontinued manufacturing, or from patient demand exceeding available supply. Having more than one manufacturing site, or many manufacturing sites, as is the case with DM, with robust quality systems and supply chains in a sponsor’s manufacturing network decreases the likelihood of a drug shortage by diversifying manufacturing locations to mitigate negative impact from natural disasters, for example, and by increasing likelihood of guaranteeing supply for and distribution to patients. Mitigating drug shortages should motivate sponsors and regulators alike to take full advantage of science- and risk-based approaches to expand manufacturing networks and available capacity, embrace emerging technologies to enable efficiencies and speed, and streamline health authority review durations; thereby, ensuring consistent, quality medicines for patients.
If regulators recognize that sponsors are “moving toward advanced manufacturing technologies, such as CM, for both small-molecule drugs and biological products... to improve the agility, flexibility, cost and robustness of manufacturing processes,” and acknowledge that, “these technologies have great potential to accelerate new, more targeted therapies, enhance product quality and bolster stability in the... drug supply...,” 51 then sponsors and regulators must embrace the submission and review of Module 3 content that is nontraditional but is science- and risk-based.
Emerging Regulatory Initiatives
The presented case studies demonstrate how existing regulations can be successfully applied to support unique manufacturing scenarios. However, adaptations to the current regulatory framework that specifically account for decentralized manufacturing models could streamline filing processes and establish a path for the implementation of novel technologies (see the enabling technologies and selected examples sections).
The use of science- and risk-based approaches and QbD should remain core tenets, but updates may help biopharmaceutical manufacturers better understand regulators’ perspectives on facility registration, risk mitigation, and process controls in the context of DM, MM, and POC. Similarly, the regulatory framework should provide advice on managing inspection requirements, site addresses, and post-transport validation when DM or POC manufacturing sites are used. Regulators in some regions are moving toward creating tailored regulations and guidance for use with DM and POC manufacturing.
MHRA
In January 2023, the Medicines and Healthcare Products Regulatory Agency (MHRA) in the UK announced its intent to develop a regulatory framework that supports POC manufacturing and issued a proposal detailing the agency’s preliminary approach to POC regulation. Prior to the announcement, the MHRA worked with stakeholders to evaluate current challenges and the need to expand current regulations with additional supportive measures for POC manufacturing. The MHRA indicated that during this evaluation period, it had seen a variety of POC products, including cell and gene therapies, small molecules, and blood products.62
The MHRA’s framework proposal outlines the importance of establishing a control site, which will be designated in the marketing application as the establishment that upholds and oversees a core PQS for a distributed POC manufacturing network.62 The control site is subject to inspection by the MHRA. It will be responsible for notifying the MHRA of significant manufacturing events and maintaining a master file. Along with the proposal, the MHRA has posed several questions to stakeholders to assess the need for and application of the proposed framework.
US FDA
Similarly, over the past decade, the US FDA has established a variety of initiatives and programs to enable novel manufacturing technologies. A recent example is the Framework for Regulatory Advanced Manufacturing Evaluation (FRAME) initiative, which seeks to develop regulatory frameworks to support innovative manufacturing technologies.63 One of the outcomes from this initiative was a discussion paper published in late 2022 to establish the FDA’s terminology list corresponding to DM and POC, as well as to provide specific areas in which the regulatory framework may need to be adapted to better fit novel manufacturing paradigms.6
The paper was published with the intent of seeking feedback from pharmaceutical developers to facilitate policy development on advanced manufacturing. The Omnibus Act, approved by the US Congress in December 2022, also influences advanced manufacturing by enabling the FDA to set advanced manufacturing and platform technology designations.64
EMA
In the EU, one of the European Medicine Agency’s (EMA’s) documents outlines its strategic goal to enable the implementation of innovative manufacturing technologies65 and to facilitate the formation of the Quality Innovation Group, which is tasked with conducting assessments, engaging with stakeholders, and issuing guidance documents on the topic of novel manufacturing technologies. The Quality Innovation Group has identified CM, DM, and POC manufacturing as key topics as part of the work plan.66
Other Regions
Though other regions have these concepts on their radar, at the time of writing, not all health authorities have announced plans to issue specific guidance or regulation related to DM, POC, or MM.
Anticipated Future Developments
Manufacturing and information processing technologies will continue to advance, expand, and influence our current ways of working to meet the needs of the evolving medical, biopharmaceutical, and regulatory environment. Under the framework established by ISPE’s Pharma 4.0™ initiative, modern manufacturing facilities are anticipated to be increasingly digitized, autonomous, and risk based by design to allow for accelerated production while maintaining robust quality standards [5].
Smart Factories
As a key benefit, “smart” factories can consistently produce large volumes of high-quality data by leveraging automation, robotic support, remote monitoring capabilities, and cloud-based data exchange. In the context of DM and POC manufacturing, such tools can enable multiple, disparate sites to be controlled under a single harmonized quality and manufacturing system, with the ability to access data from anywhere via cloud-based enterprise systems. In this digitized environment, both regulatory submissions and health authority inspections can be made easier because data can be quickly harnessed and compared across multiple sites without the need to manually compile or physically visit individual locations.
Next-Generation Knowledge Management
With an increase in the overall volume of data generated, pharmaceutical manufacturers will also need to apply specific solutions to help manage, store, parse, analyze, and mobilize data in the context of knowledge management.67 Additionally, approaches in structured content management, as well as artificial intelligence (AI) and machine learning, can help make data accessible and usable in downstream applications, such as regulatory filings.
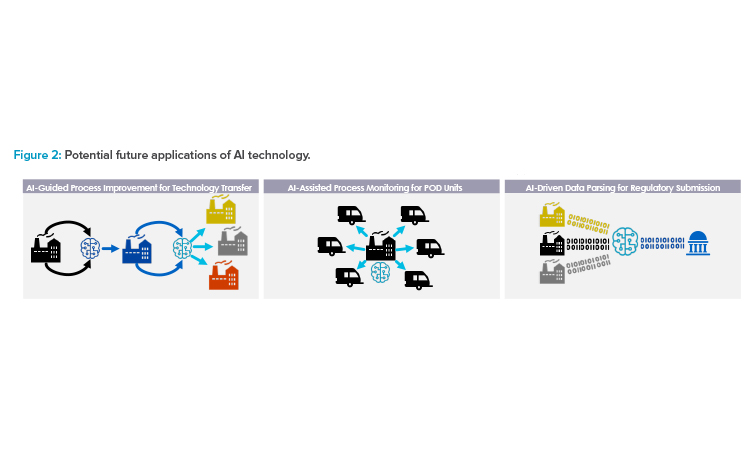
Although next-generation AI toolsets, such as text mining and natural language processing, can process unstructured data, having a structured data model helps AI tools work more efficiently and ensures the captured data is usable and kept in context with its source (see Figure 2). Layering AI tools, such as large language models (e.g., OpenAI’s ChatGPT and others), on top of a structured data model can allow for further extensions in capabilities.
Cloud-Based Data Exchange
Cloud-based platforms will also help foster digital connectivity across physical locations. In particular, a cloud data exchange solution that connects sponsors and health authorities for real-time data exchange would help ease the burdens of continuous, high-volume data flow. Accumulus Synergy, a nonprofit founded in 2020, is developing a cloud platform with collaborative capabilities that can help reach this vision.67
Dynamic Real-Time Approaches
Adapting more dynamic real-time approaches to analytical testing and feedback control, such as PAT, will become a crucial element of a cohesive control strategy that can be applied across different facility locations. In a future scenario, data could be streamed to a remote monitoring site and evaluated in real time, enabling feedback controls to ensure in-specification quality attributes and allow RTRT (see Figure 2). Additionally, manufacturers may be able to more rapidly adjust and modify conditions or procedures at one site based on testing outcomes from another site. Conversely, in the case that there is a failure in one site, AI learning may be leveraged to reduce or prevent the risk of failure in another site.
Simplified Technology Transfers
Extensive data collection, integration, and AI tools may also be used to model specific trends and develop predictive models that can inform the change management process. With necessary changes to the regulatory framework and an increased understanding of AI application and deployment, AI models could simplify the technology transfer process.
For example, if a sponsor uses a traditional approach for two or three initial technology transfers and adapts an AI algorithm to “learn” how to mitigate risks incurred during the process, subsequent technology transfers could be conducted under a “low” risk assessment assumption, as deep scientific understanding is significantly increased through AI learning during the previous technology transfer processes (see Figure 2) based on which changes are guided and executed.
AI Technology
There are multiple potential future applications of AI technology that can support more efficient technology transfer, scale-up, site monitoring, and regulatory submission processes (see Figure 2). AI can guide process improvement through iterative risk evaluation, wherein changes are made in response to an AI algorithm’s readout, with data obtained using dynamic methods such as PAT. This can simplify future scale-up and technology transfer. Similarly, AI applications could be used as part of a remote monitoring strategy for multiple different decentralized sites, such as POD units. Finally, AI can assist with managing large volumes of data by enabling automated data parsing and sorting to streamline regulatory submission preparation.
Autonomous Production Systems
Some of the unique challenges of POC manufacturing, such as the training and qualification of on-site personnel, can be remedied by onboarding fully autonomous production systems. Healthcare workers are typically not trained in GMP manufacturing and are therefore not qualified to operate most POC facilities. If end user operation at the healthcare facility is extremely limited, fully autonomous production systems with virtual control may reduce the burden of personnel limitations.
Regulatory Considerations for AI Tools
Regulatory considerations for applications of AI and machine learning is an area of active discussion within several health authorities because greater understanding is needed before policy can be formally issued.66, 68 AI has the potential to introduce significant complexity into manufacturing decision-making processes, which has downstream impact on regulatory assessment. As a result, a more realistic, near-term strategy may be to utilize AI to assist with data mining and text generation to streamline the regulatory submission authoring process, which will be necessary in the context of multiple sites.
Conclusion
Without a supportive regulatory framework, advances in biopharmaceutical manufacturing technology and innovative operating models will be impractical for sponsors to implement globally. Although technology and regulation have traditionally been on very different timelines, with technological innovation developing at an ever-increasing pace and the resulting regulations slow to follow on, they must ultimately evolve together to be maximally effective.
A balance must be reached to ensure that the biopharmaceutical industry can take full advantage of what the technologies have to offer and meet the challenges posed by the ever-changing supply and demand situation, as well as the evolving landscape in pharmaceutical manufacturing. Notably, patients are the ultimate benefactors of this evolution, as they will have more ready access to medicines globally.
Acknowledgements
The authors would like to thank Mike Abernathy, Jette Wypych, Inna Ben-Anat, Andrew Lennard, Twinkle Christian, Jackie Milne, Tony Wang, Pei-Shan Yen, Venkatesh Natarajan, Simon Hotchin, Chetan Goudar, Randy Mohr, Scott Siera, and Yusdy Pan for their review and comments.






