Toward a Holistic Control Strategy: Bridging Gaps Across the Product Lifecycle

This article defines what constitutes a holistic control strategy in pharmaceutical development and manufacturing. It highlights the limitations of traditional, siloed approaches and demonstrates the added value of integrating these approaches across multiple dimensions. By identifying key gaps and proposing technological enablers, the article outlines a path toward a more comprehensive, lifecycle-spanning control strategy that enhances product quality, process performance, and regulatory flexibility.
A control strategy (CS) is a planned and structured set of measures—such as tests, monitoring, and controls—based on a thorough understanding of the product and its manufacturing process, which are designed to ensure that the process consistently performs as intended and that the final product meets quality standards. The CS is the essential outcome of process characterization and phase 1 process validation.
However, current approaches often reveal significant gaps during the development and deployment of the CS. These gaps arise because different aspects of the dimensions, such as the product life cycle, the process chain (including the supply chain for sourcing Active Pharmaceutical Ingredients and excipients), the facility design, and clinical studies—are not combined.
This article aims to propose good practices and technological enablers to achieve a holistic control strategy defined as one that encompasses all relevant dimensions. We want to show that the CS can be structured into different dimensions and the interactivity between the different dimensions of the CS will leverage substantial business benefits. First, we address good practices of each dimension to resolve gaps. Then, we propose cross-dimensional technological enablers and their requirements, and conclude by outlining the potential benefits of adopting a holistic CS vision.
INTRODUCTION
The CS has always been a requirement but was historically mainly interpreted by end-product testing and narrow control of manufacturing processes and materials without a comprehensive understanding of the relationships between process parameters, material attributes and product quality attributes. The CS concept has evolved with the implementation of ICH guidelines Q8 to Q121, 2, 3, 4, 5in the past 15 years. A new pharmaceutical paradigm has emerged that uses Quality by Design (QbD)1 as a strategic framework, highlighting science and risk-based approaches along with product and process understanding. In an enhanced approach to manufacturing process development using QbD concepts, an enhanced CS is based on a better understanding of the product and the process, which allows identifying the material attributes and process parameters that should be controlled. Scientifically sound, data-driven, and based on risk assessments, this approach allows the CS to focus on fundamental elements influencing the quality target product profile (QTPP) and critical quality attributes (CQAs) and ensures that product quality is met in a predictive manner.
The control strategy, derived from current product and process understanding, is built up throughout pharmaceutical development (stage 1 of process validation) and becomes a central repository for regulatory filing. It is also the reference to ensure product quality and process performance (stage 2) during commercial manufacturing and the product life cycle (stage 3, Continued Process Verification (CPV) and change management).
Building an efficient CS for the commercial phase of the product, relying on the basics of the product and process risks, appears even more necessary and relevant as ICH Q125 has been published to facilitate change management during the product life cycle, which encourages innovation and continuous improvement.
Definitions
The definition of the control strategy, introduced in ICH Q103, highlights that the CS contains several levels, and the understanding of these different levels and their role in the CS is crucial for product life cycle management. The ICH Q10 definition of control strategy is “a planned set of controls, derived from current product and process understanding, that assures process performance and product quality. The controls can include parameters and attributes related to Drug Substance (DS) and Drug P\product (DP) materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control”3.
In this definition, one level of the CS is focused on the product-oriented elements (i.e., finished product specifications) and the associated methods and frequency of monitoring and control. The second level represents process-oriented elements (i.e., parameters and attributes related to DS and DP materials and components). This includes supply chain transparency and control. The third level contains the operations-oriented elements through facility and equipment operating conditions. The starting point of these different levels is to satisfy the patients’ requirements, i.e., the QTPP.
The product-oriented elements are associated with analytical methods, being themselves subject to a development strategy and an analytical CS to monitor their performance throughout the method life cycle. Within the process-oriented elements, materials and components require material and process characterization to identify which material attributes interact with process parameters to influence the product quality. Materials and components also need a robust pharmaceutical quality system (PQS) throughout the supply chain to assure that when the critical and key material attributes are identified, they are effectively met by all suppliers.
The operations level is deployed through facility design, equipment and information systems design, definition of data to be recorded and monitored, procedures implementation, people training, to assure the product and process levels of the CS are correctly implemented. This “operations” level of the CS is implemented on each manufacturing site, whereas the product and process levels should be aligned between sites, as highlighted by ICH Q11 5: “the knowledge and process understanding should be shared as needed to perform the manufacturing process and implement the CS across sites involved in manufacturing the drug substance.” Nevertheless, these different levels are not always well connected, and gaps can be observed in the development and deployment of the control strategy.
GAPS IN THE DEVELOPMENT AND DEPLOYMENT OF CS
As mentioned, it must be understood that a CS is not only set up once—a life cycle capability must also be enabled. Are control strategies established in a way that is sustainable for other stages of the life cycle? Sometimes it seems they are only built for archiving systems. What are the requirements of models being applied in later stages, e.g., validation of models for usage in manufacturing?
The crucial role of the CS during the product life cycle is reinforced by ICH Q12 guideline5, which provides a framework to facilitate managing postapproval changes and affects the Chemistry, Manufacturing, and Controls (CMC) part of the regulatory dossier. Using enhanced product and process understanding, the industry will be able to reduce the number of regulatory submissions and implement these changes in a more efficient and predictable way. To allow these benefits, the control strategy plays a central role in the ICH Q12 approach to identify and appropriately justify the established conditions which are the elements considered necessary to assure product quality and which will trigger a regulatory action if changed. This also identifies and appropriately justifies the non-EC that will be allowed to be managed through the PQS.
Because the life cycle idea is rarely employed and because setting control strategies is still considered a one-time effort, we analyzed the gaps of the currently interpreted CS along the different stages of the product life cycle.
| Lifecycle Stages | ISSUES AND CONCERNS |
|---|---|
| Pharmaceutical Development |
|
| Clinical Trials |
|
| Technology Transfer |
|
| Commercial Manufacturing |
|
| Organizational aspects throughout the lifecycle |
|
Motivation and Goal of this Contribution
As it can be extracted from the gap analysis above, the CS needs to cover and combine multiple dimensions, which we want to define as follows:
- 1. Achieve understanding along the process chain, influence of raw / starting materials, hence also supply chain control, from upstream- and downstream manufacturing, DS, DP, to the patient. Including leveraging knowledge across products as platform knowledge
- 2. Transfer process understanding along the product life cycle from development to manufacturing
- 3. Interweave clinical study design and execution with process validation stages
Feed process understanding into the engineering activities of facility design and use specifications for process validation
Those dimensions are depicted in the following figure, on which this contribution focusses.
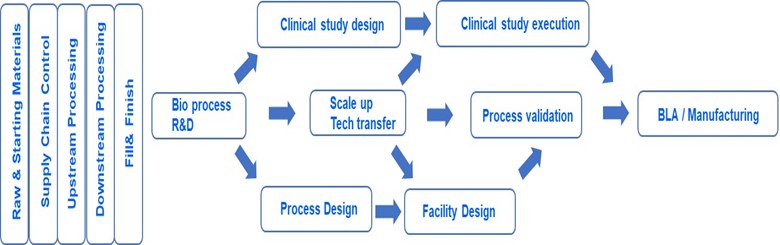
Thus, in this article, we propose good practices and technological enablers to achieve a holistic (i.e., covering all dimensions) control strategy. Further, we aim to show that a control strategy can be structured into different dimensions, and interactivity between the different dimensions of the control strategy will leverage substantial business benefits. We will first address good practices of each dimension to overcome the gaps already mentioned and subsequently interweave the dimensions by proposing cross-dimensional technological enablers and their requirements, then concluding with the potential benefits of the proposed holistic control strategy vision.
Comparison With Other Approaches
Advancing Pharmaceutical Quality (APQ)™
The maturity level of control strategies was assessed in ISPE’s APQ initiative6. The APQ maturity model checks the maturity of the CS at each stage of the life cycle and mentions a few required capabilities, such as analytics and development approaches and the general need for digitalization and data analytics. However, it did not have the goal to address the holistic nature of the control strategy, including the following:
- Connect the different unit operations
- Connect the different stages of the product life cycle
- Respect the CS in the facility design phase
- Interlink process control to clinical study impact
The APQ initiative addresses the maturity of the control strategy and how elaborated the CS is, but it does not aim to address the holistic interconnectivity between the dimensions previously outlined.
ICH M4Q
The ICH M4Q guidelines suggested an overall CS in the respective concept paper7. To enable this, a revision of the common technical document (CTD) structure is necessary. To focus on the overall CS concepts, comprehensive description of the control strategy and implement fully the tools and enablers introduced in ICH Q12. The concept paper introduces the terminology of “overall control strategy” and suggests that it should be the backbone of the revised M4Q structure.
This term will need to be defined in the revised guideline, but we can anticipate that it contains end-to-end, holistic considerations from raw and starting materials to the final DP, encompassing all materials and steps involved in the manufacturing process of the application. A comprehensive representation (graphical, tabular representations with cross reference to other CTD sections, etc.) would be very helpful to facilitate the understanding of the key elements assuring the product quality, from the applicant side and the evaluation side. Adoption of the final guideline is planned for November 2026. The goal of this overall CS is to provide an overall vision of the connections between all the CMC elements provided in the CTD and interacting to meet the QTPP. Hence, the CS must be correctly designed and described in the regulatory file, i.e., the new drug application (NDA) and biologic license application (BLA). This will allow regulatory assessment and marketing authorization acceptance as well as product lifecycle management and change management (to avoid regulatory burden).
PROCESS AND PRODUCT DEVELOPMENT AND CHARACTERIZATION
For both product- and process-oriented control elements, data-based evidence should be presented in terms of process validation to demonstrate the ability to consistently deliver product quality8, 9 . Moreover, both follow a risk-based approach that starts with a risk assessment to rate quality attributes based on the product’s mode of action as well as the criticality of process failure modes and related controllable process parameters. This is necessary because the impact of all process parameters on quality attributes cannot be evaluated experimentally in economically feasible boundaries.
Product-oriented elements
Controls related to the product-specific attributes will be referred to as specifications at the DS and DP level and as in-process-control limits for product quality limits at intermediate process steps. Having quantitative understanding and justification of specifications and in-process-control limits is key to an enhanced approach and to bringing QbD principles to life. Product-related control strategies, such as end-product specifications and in-process controls, are still commonly defined using x-SD approaches based on limited development or clinical data. This often results in overly broad specifications, which weaken process control, or overly narrow ones, which penalize well-controlled processes. Additionally, these strategies are typically not aligned with patient needs. Because usually three standard deviations is used to set these product-related control limits, 3 sigma process can be enabled at maximum. This may lead to either too narrow specifications, putting drug supply at risk, or to too wide specifications, putting patient safety at risk.
Establishment of patient-driven specifications and in-process-control limits needs to be viewed as a must-have and not a nice-to-have if established controls should meet patient needs, not process needs. Examples on how to establish more patient-centric specifications are given in an industry-wide publication, “Strategies for Setting Patient-Centric Commercial Specifications for Biotherapeutic Products”10.
Setting specifications is handled in ICH Q6B11. Setting patient-centric specifications is a topic on its own and not addressed in depth in this article. For the rest of the article, it is assumed that clinically relevant specifications can be obtained, e.g., via in vitro/vivo studies, prior knowledge, animal studies, or cell-based assays up to human-on-a-chip technologies.
With such approaches, a patient surrogate model can be established that links CQA variability to impact on clinical efficacy and safety. Although representativeness of such systems with respect to clinical studies conducted in humans needs to be understood, we need to compare it to the status quo. This is comparable to gambling, as we take 3–5 standard deviations of a very small population, usually around 3–5 runs, of clinical batches to derive specifications and in-process-control limits. Any scientifically undermined approach would usually outperform by taking standard deviations from 3–5 runs, which rewards for having large process variability among those 3–5 runs.
Summarizing, without having justified and scientifically sound patient-centric specifications and in-process controls in place, any development of a CS on process parameters is meaningless.
Process-oriented control elements
Process models are key enablers for data- and risk-based decision-making on control ranges for process parameters and material attributes. All process models share a similar form, as they predict product quality and/or product amount as a function of process parameters and material attributes. Hence, they can be used to infer levels of process parameter and material variability to still achieve sufficient levels of product quality and product amount, taking model and sample uncertainty into account. This is a major benefit over relying on a very small (minimum n = 1) set of experiments that impede a scientifically sound risk assessment of process parameter variation on product quality.
Varying levels and implementations of control strategies exist, such as static control ranges (e.g., proven acceptable ranges [PARs] or design spaces) or more advanced, adaptive control strategies that run even in closed-loop form. Still, most process parameters and material inputs are controlled via control ranges. Adaptive controls are still rare to be found adopted throughout the entire life cycle, e.g., employed in a manufacturing environment. Reasons are multifold, but usually it is driven by the fast track of development, reduced frontloading of advanced technologies until market launch, and subsequent reluctance to change.
Control ranges—e.g., PARs or design spaces—serve as guidance during manufacturing because operators and equipment are trained and selected to stay within those boundaries. As a misspecification of those control ranges might lead to a systematic decrease of process robustness, setting those control ranges, which constitute of a set point as well as a range around, can been seen as a critical task within the QbD framework. New regulatory guidelines aim at harmonization of used process models and focus on model validation and verification 12. Established control ranges and conditions must be filed in a dossier, as they could be later used to justify postapproval changes, e.g., using design spaces.
- There is a trend for “technology platform” (multiproduct) possible submission vs. (single) product submission. Although each dossier that describes the CS for the individual product needs to stand on its own, it is regulatory accepted and promoted to include prior knowledge of previous, similar products to foster the development of new products. Nonetheless, a scientifically sound approach needs to be chosen when incorporating prior information. Bayesian approaches might be particularly useful when referring to information on control strategies created in prior, similar product applications.
In summary, a holistic description of product-oriented and process-oriented control strategies that enable risk-inferred decision-making using scientifically sound models, including uncertainty quantification, can speed up later stages of the product life cycle, such as shortening postapproval changes, deviation management, and advanced process control.
CONTROL STRATEGY IN SCALE UP AND TECHNOLOGY TRANSFER
Demonstration of similarity and transferability of control strategies is a frequently visited task during the product life cycle. This may happen between scales, across manufacturing sites, or across companies, e.g., when moving from one Contract Manufacturing Organization (CMO) to another. Depending on the level of understanding of the CS as a function of potential changes it might be possible to transfer knowledge of the existing CS to a new setup.
However, because nearly all models that describe the impact of scale, manufacturing equipment, etc. on quality attributes use empirical relationships, there’s a small risk when introducing changes. During process characterization, it is essential to define a CS that can be transferred or ruled out on different scales and/or manufacturing sites. Experiments may span over multiple scales and may include multiple equipment that can be characterized upfront. Although, due to tight timelines toward time to market, this is not done frequently.
During technology transfer, the process is translated to the commercial scale. It is at this stage that the CS is usually finalized, requiring another round of process risk analysis focused on industrial-scale impacts. This time, the Critical Process Parameter (CPP) and Critical Material Attribute (CMA) identification is already performed: the goal is to identify how they are controlled at industrial scale and with the technologies in place in the plant. This part of the CS is often called Process control strategy. Hence, it is recommended to perform a gap assessment between scales and manufacturing sites to ensure that the CS “high level” is preserved.
Assess the level of impact of the changes on product quality and process performance and identify action plans to reduce the risk (development/characterization studies, pilot-scale batches, industrialization/engineering batches) to confirm that the CS is adapted to real conditions and at industrial scale. This illustrates that the holistic control strategy is a dynamic element, with its own life cycle, and that it must be adapted, driven by changes, transfer, and other events through a product life cycle.
Clinical Studies
Best Practices
Implementing effective control strategies in clinical studies is essential for ensuring process consistency, regulatory compliance, and successful study outcomes. The following practices can help establish robust control strategies during clinical trials.
- Early risk assessment: Identify and prioritize Critical Quality Attributes (CQAs) and CPPs early in clinical development. Risk assessment tools, such as matrices and Failure Mode and Effects Analysis (FMEA), support proactive identification and mitigation of risks, helping ensure patient safety and data integrity.
- Robust process design: Use small-scale and scale-down models to simulate manufacturing conditions for clinical trial materials. These models allow for testing and optimization of process parameters to identify potential sources of variability before full-scale clinical production.
- Standardized protocols: Establish clear, standardized workflows for batch documentation, deviation management, and change controls. Consistent execution of these protocols ensures data reliability, traceability, and compliance with regulatory guidelines.
- Real-time data monitoring: Implement process analytical technologies (PATs) and real-time monitoring systems to track CPPs and CQAs during clinical manufacturing. This approach enables rapid detection of deviations and immediate corrective actions to maintain product quality.
- Data integration and feedback: Integrate manufacturing data with insights from PD to support continuous improvement. Use data-driven process reviews to refine control strategies, address variability, and enhance process understanding throughout clinical phases.
By following these good practices, biopharmaceutical companies can ensure that clinical studies are executed with precision, meeting quality standards and regulatory expectations while supporting the successful development of new therapies.
CQAs and Clinical Results
A well-known hurdle is successfully showing efficacy in clinical trials. According to industry analyses, around 90% of drug candidates fail during clinical trial phases 1–3, and 50% of failures are due to insufficient drug efficacy23. This highlights a significant opportunity for manufacturers—especially in the biopharmaceutical space with complex CQAs—to leverage process knowledge to fine-tune production in ways that could enhance clinical efficacy and reduce trial failure rates.
A long-missing connection is between the CQAs and their clinical impact. This has many symptoms, such as the inability to define what are CQAs for certain products like Advanced Therapy Medicinal Products (ATMPs), or to derive clinically relevant (i.e., patient-driven) specifications. Due to the upcoming advanced clinical surrogate assays, like organ or patients-on-a-chip, identification of CQAs and their relevant ranges (i.e., specifications) is critical to the optimization of the process to ensure those will be possible.
Therefore, the holistic control strategy shall interweave clinical data study planning and its outcomes (i.e., efficacy) with the stage 1 process characterization work (CQAs) to focus on the link between CQAs and clinical results. This is a new era of targeted PD to patient needs, which has the potential to significantly increase clinical success.
This requires not only new equipment, but also modeling tools that enable linking process understanding to expected patient outcomes. Although concerns on the representativeness of clinical surrogate assays may be raised and further improvement of those technologies will happen in the future, a comparison shows that this approach is more systematic and targeted to success. State-of-the-art is to use a very limited amount (e.g., 3–5) of clinical batches to test in clinical trials. Depending on the process CS the quality might be less or more consistent across those batches. However, the optimal CQA profile is not known given the boundaries of manufacturing. We can compare this to testing out one CQA fingerprint out of a million possible combinations of CQA pattern.
CS in Manufacturing
Establishing an effective CS is critical for ensuring consistent product quality, process efficiency, and regulatory compliance in pharmaceutical manufacturing. The following methods provide a comprehensive approach to achieving these objectives. By integrating these methods into a holistic control strategy, manufacturers can ensure robust processes, drive continuous improvement, and deliver high-quality biopharmaceutical products efficiently.
Golden Batch
To achieve the desired performance, it is essential to first identify sources of variability at the manufacturing scale to understand and control factors that impact process outcomes. Then, stabilize performance at the golden batch level, ensuring the lowest batch-to-batch variability by aligning process conditions and outcomes with an optimal reference batch. Finally, determine set points for CPPs within their PARs, allowing for robust and repeatable manufacturing.
Process Optimization
Once golden batch stabilization is achieved, productivity can be further optimized while maintaining product quality. Adjust and refine the CS multidimensionally within the design space. This approach avoids the need for postapproval change submissions while enabling process adaptability. Then, leverage process understanding derived from PCS to simultaneously optimize multiple CPPs. Use scale-down models and targeted manufacturing runs to validate slight process variations and their impact on productivity and quality.
Continuous Improvement Beyond CPV
- CPV should be viewed as a foundation for ongoing enhancement, not just a compliance activity. Move beyond univariate trending of CQAs and Key Performance Indicators(KPIs) with traditional statistical limits (e.g., Nelson rules). Incorporate novel manufacturing data aligned with ICH Q12 guidelines to continuously improve processes. By merging manufacturing, PD, and PCS data, the CS can evolve in a traceable and effective manner both inside and outside the design space.
Out-of-Specification Handling
Efficient Out-of-Specification (OOS) management relies on a data-driven approach. Integrate data from PD, PCS, and manufacturing operations to identify root causes holistically rather than relying solely on manufacturing data. Accelerate root cause identification using a contextualized, holistic data management platform that streamlines data analysis. Implement advanced data analytics and hybrid models to create platform knowledge, fostering a knowledge-based system that enhances decision-making.
Real-Time Release
Real-Time Release Testing(RTRT) enables faster product disposition through process understanding and predictive analytics. First, develop and validate end-to-end digital twins across the entire process chain to demonstrate suitability for real-time use. Then, deploy the digital twins for predictive analytics, enabling data-driven recommendations that optimize pending unit operations and ensure batch success. Once complete, implement recommendations seamlessly via electronic batch records integrated into the manufacturing execution system (MES), allowing direct adjustments of control set points to enhance outcomes.
ICH Q12–Oriented Adaptation, Post Submission
CS is builtup throughout pharmaceutical development and then becomes both a central repository for regulatory filing and the reference to ensure product quality and process performance during commercial manufacturing and product life cycle (CPV, change management). This statement will be even more reinforced by the planned revision of the ICH M4Q guideline.
With the adoption of ICH Q12 guideline, implemented in some ICH regions (e.g., US, Japan, China) and in the process of implementation in others (e.g., EU), the CS and the way it is described in the regulatory file has become even more crucial to facilitate change management during the product life cycle, and thus encourage innovation and continuous improvement.
The way the CS is designed throughout the development phases is crucial for the extent of ECs that will require a regulatory submission (i.e., variation) in case of change. Increased knowledge and understanding will allow the justification of non-EC (information considered not necessary to assure the product quality) that will be managed through the PQS of the marketing authorization holder (MAH).
Increased understanding of the interactions between inputs (process parameters, material attributes) and product quality will limit the number of ECs that will focus on the most important parameters, material attributes, and outputs, if appropriate specific in-process controls (IPCs). On the contrary, limited understanding and limited development studies will increase the extent of ECs, including many inputs together with outputs. Therefore, the effort put in the development phase will be leveraged later during the product life cycle management, reducing unnecessary cost and time burdens for regulators and industry on low-risk changes.
Cross-Dimensional Technological Enablers and Requirements
This section proposes tools and their requirement to interweave the individual dimensions of the control strategy.
Cross-Dimensional Risk Assessments
To build an evolutive and efficient holistic control strategy, risk assessments are performed throughout the life cycle. These risk assessments have a specific purpose at each life cycle phase. The approach can evolve because the knowledge is growing, the inputs for the risk assessments are increasing, and the outputs are used for different purposes.
- Product development: Product risk assessment assesses the criticality level of quality attributes regarding safety and efficacy, identify the uncertainty, and identify studies and knowledge to gain.
- Review and populate the risk assessment with the knowledge gained with the studies (in vitro, clinical data, etc.).
- PD: Process and materials risk assessments assess the criticality level of process parameters and material attributes, identify the missing information, and prioritize the studies to perform to confirm the impacts, the level of control needed, and define the acceptable ranges (process parameters) and acceptance criteria (material attributes).
- Scale up and/or tech transfer: To prepare a tech transfer or major process change, a specific risk assessment is needed to compare the process on all its dimensions (i.e., process parameters, materials used, process conditions like environmental, equipment and technologies, in-process controls and measurements), identify the differences, and assess their impact on the product quality and process performance. According to this impact analysis, action plans are identified to secure the risk through small-scale studies, technical batches, specific testing on equipment, process validation, and the final demonstration of comparability.
All risks assessments are performed by a human operator, generally with conventional tools like Microsoft Excel spreadsheets and Word documents, and require the collection of a significant amount of information, data, and documents. These conventional tools propose poor visualization solutions, whether three dimensions (product, process, materials interactions) would be necessary.
In the end, although a huge workload and effort has been made to reconcile all this information in the risk assessments tools, risks of discrepancies and redundant information still remain and the maintenance of these tools is still a burden. This mine of information remains a static knowledge, not efficiently shared and is finally wasted.
Digital tools are arousing hope to provide solutions for the gaps detailed previously. They can provide solutions to accelerate data collection, analysis, and entry as input data for the risk assessments; enhance consistency; and enhance knowledge-sharing across company departments and sites. They can also provide powerful visualization tools that will enhance product, process, and materials linkage understanding. With digital tools, it may be possible to finally enable a real life cycle vision and forecast of the control strategy.
The development of these tools, supported by artificial intelligence (AI) technologies, is progressing very fast and will certainly provide robust solutions soon to support people in the health industry and to help them spend their time and energy on value-added tasks and decrease the workload on documentation generation and verification.
Data Management, Process Automation, and Data Analytics
Data management, process automation, and data analytics are essential tools for establishing robust holistic control strategies in biopharmaceutical manufacturing. A CS is not static; it requires continuous evaluation across different time scales, both offline and in real time, to ensure process reliability, adaptability, and compliance.
Offline applications
- Offline applications should be evaluated during CS development in PCS, in which data management supports the systematic identification and optimization of CPPs and CQAs. They can also evaluate trending analysis in CPV, where automation and analytics enable the identification of long-term trends in process performance, ensuring alignment with ICH Q12 guidelines for continuous improvement. Finally, combining manufacturing data with insights from PD allows for ongoing optimization and continuous improvement of the control strategy and adaptation to new data sources.
Real-time applications
Real-time data analytics enable precise decision-making for transitions, such as the timing of harvest or pooling decisions, which are critical for product quality. Automation supports closed-loop control by adjusting process parameters in real time based on continuous feedback. (This will require digital twins, as addressed next.) With informed control blocks, advanced analytics fuse data sources to enable predictive, dynamic adjustments to the control strategy.
Integration of data sources
To ensure a comprehensive and holistic control strategy, data must be integrated across all automation levels. In level 1 and 2, SCADA systems and PAT provide critical process data but are not sufficient on their own. In level 3, MESs and laboratory information management systems (LIMSs) add product quality data to enable broader analysis. In level 4, integration with supply chain and market data allows for adaptive control strategies that align manufacturing with market needs. The control strategy links CPPs and CQAs, requiring data from all these sources to be fused into a unified system. The control strategy itself operates outside of these levels but connects them seamlessly, enabling robust process control and continuous improvement.
Automation, architecture, and algorithmic needs
For automated data collection, implementing data import architectures and bidirectional interfaces ensures efficient, real-time input into the CS. Testing these interfaces for data integrity is critical, even in nonvalidated environments. Activities requiring validated environments must be clearly defined to ensure compliance while allowing for development in nonvalidated spaces. Deterministic algorithms such as digital twins enable automated calculations and simulations at different time scales, supporting real-time decision-making and offline analysis.
By integrating PCS, PD, and manufacturing data, a robust, adaptive CS can be developed and continuously improved. Automation, data management, and advanced analytics form the backbone of this approach, ensuring biopharmaceutical processes are efficient, compliant, and resilient to variability (see Figure 2).
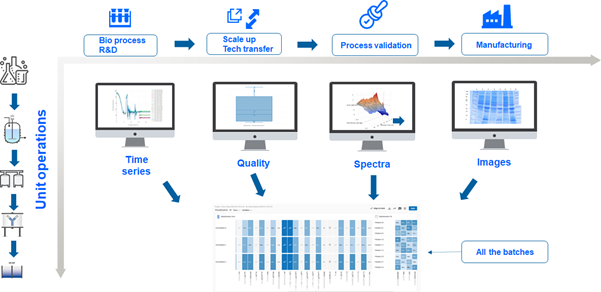
This approach—exemplified on the two dimensions of product life cycle management (PLM) and the unit operations—of course also holds for the further dimensions such as facility design and clinical data. Hence, we encourage establishing a “data link” vision to be able to link all the elements of the control strategy to the facility design activities as well as to and from clinical study design and execution.
Cross-Dimensional Process Models and Digital Twins
An important idea is to understand all manufacturing processes as a multi-input multi-output (MIMO) system. Multiple unit operations, each having multiple controllable inputs such as process parameters, are producing a product that is characterized using multiple CQAs as well as the product amount. Moreover, it is critical to understand that selling manufacturing batches or units is only possible if all (and only all) specifications are met.
In the case that we neglect that complexity for modeling, the impact on the CS and subsequent impact on business benefits is not evident or is even misleading. Simple examples for such mismatches are modeling of only product amount or only a subset of CQAs, which might lead to a high yield but low quality. Also only focusing on optimization of a single unit operation might lead to loss of product or even unwanted effects on other unit operation not buying into the global objectiveness. Nonetheless, process models have been predominantly established per process step individually, forming silos of information and in certain cases, focusing only on a subset of outputs.
In contrast, interdimensional, holistic, and end-to-end process models connect along multiple dimensions. The first is along the product life cycle: Process models should be able to cope with data from different scales and stages of the product life cycle, which requires that data be designed in an uncorrelated manner to combine these data sets. (This has been demonstrated recently13.) Second is along the process steps: process models should be able to predict impact of all relevant process steps in final product quality. These process models also connect with data science approaches to correctly translate knowledge from site to site or scale to scale, which includes tasks in facility design and clinical studies. Process, product, and clinical impact are related and need to be modeled together.
End-to-end process models are a frequently used and established tool to calculate in-process-control limits that ensure meeting specifications at a certain probability (see Figure 3). Hence, they provide a scientifically sound way to link in-process-control limits to specifications and overcome arbitrary setting of x-SD boundaries, which are not linked specifications at all 14, 15. Digital twins, when combined with machine learning (ML) and AI, facilitate self-adaptive control strategies. These advanced tools enable full process learning, aligning with the life cycle needs outlined in ICH Q12.
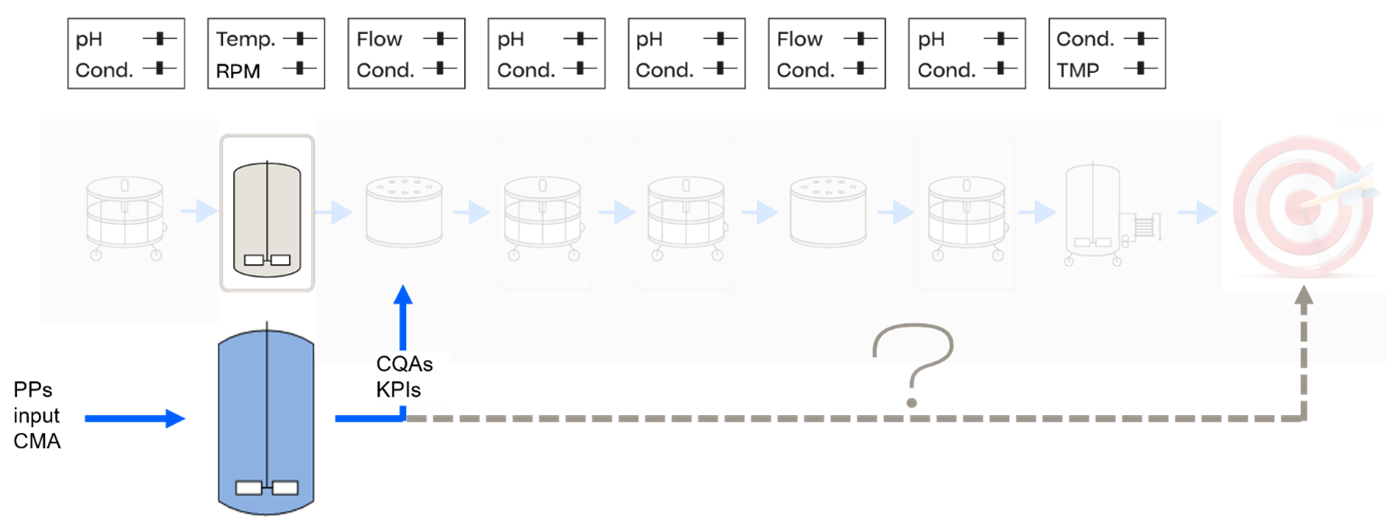
A fully connected end-to-end process model deployed as a digital twin covers previously listed dimensions. This requires the following prerequisites.
Data and validation
Data needs to be designed from the beginning of the product life cycle to be part of a final holistic control strategy. This can be achieved by paying attention to the correlation structure and designing linking experiments that ensure interaction between existing process knowledge and newly created experiments. To achieve transfer learning across multiple modeling and experimental attempts, it is required to ensure that those basic principles are met. The validation of the digital twins and end-to-end process models is, of course, different from the validation of a control strategy. Validation of digital twins can be performed within existing frameworks 16, 17].
Modeling, automation, and offline applications
- It’s important to understand the necessity to model the correct inputs and outputs, e.g., experiments measured only on a subset of the outputs need to be repeated otherwise. Automation to model building and validation is also key, as this becomes an iterative task of the life cycle and cannot be viewed as one-time exercise. The holistic CS will need digital twins not only for real-time application and autonomous adaptation, but also for offline applications such as model-based design of experiments for optimization setting multivariate PARs and normal operating ranges (NORs) inside of the design space.
Organizational Changes
As can be extracted from earlier analysis, we need to address multiple organizations at the same time, such as manufacturers, development groups, CDMOs, analytical labs, and clinical trial organizations. This article primarily addresses the head of PD, head of digitalization, head of process validation, and responsible persons in CMC regulatory functions. But this may also need to be extended to include supplier organizations because of prequalification of raw materials as well as to equipment suppliers to include equipment design.
Different disciplines need to be coordinated tightly together to design and implement the holistic control strategy along the product life cycle. As an organizational proposal, we propose a dedicated role tasked to establish a cross-functional approach to control strategy, which is currently missing, and to oversee the dimensions of the control strategy.
Due to the multidimensional nature of the CS, issues in knowledge transfer, sharing, and management arise. This is fundamental for product and process knowledge management. We need more software solutions and business processes that follow ICH Q10 to share the knowledge provided by the CS more efficiently across phases of pharmaceutical development and throughout the product life cycle.
Therefore, we want to highlight the need to develop tools for a holistic CS, as well as a tool to manage or dashboard the status. Although comprehensive quality risk management (QRM) tools are available, this should include knowledge management (KM) functions (ICH Q10)18, 19]. QRM and KM are enablers to build the control strategy. But, on the other hand, the CS is the cornerstone of QRM and KM during the commercial phase.
Those KM tools should at least feature:
- Automated digitalization in a life cycle mindset to enable knowledge that is always up to date
- Harmonized file formats across organizations, departments, and CDMOs
- A transferrable CS by deriving scale-independent parameters
- The ability to include current and future technology stacks
- Visualization of the links between CPP/raw material attribute (RMA) and CQAs
- Interactive and collaborative analysis tools
CONCLUSIONS
There were initiatives to develop a holistic CS in the past20. However, the difficulty to understand “holistic” and how to apply “holistic” to real-world product life cycle tasks was pending. First, we need to account for the fact that we are dealing with a MIMO system. The multiple outputs are the long list of CQAs, which need to be met, all of them at once and predictively. The multiple inputs are all process parameters, raw material attributes along the process chain and along the product life cycle, but also facility design aspects, clinical study material, etc. Hence, to meet all outputs, we also need to control all inputs. Only when taking this into account can we can meet the overall objective of pharmaceutical manufacturing. Thus, the term “holistic” is to address this full MIMO problem: determine the different dimensions and find overlaps between them.
The holistic CS is the connection between dimensions and enablers, helping ensure an optimized and robust CS for pharmaceutical processes. These four dimensions collectively lead into a holistic control strategy: process chain, product life cycle, clinical studies, and facility design (see Figure 4). We propose to deploy at least the following technological enablers: a risk assessment, data management, digital twins, and organizational changes. A holistic control strategy is dynamic. It has to be iteratively adapted to all changes of a product, process life cycle, etc. Business workflows that follow ICH Q10 and Q12 need to be established for this iteration.
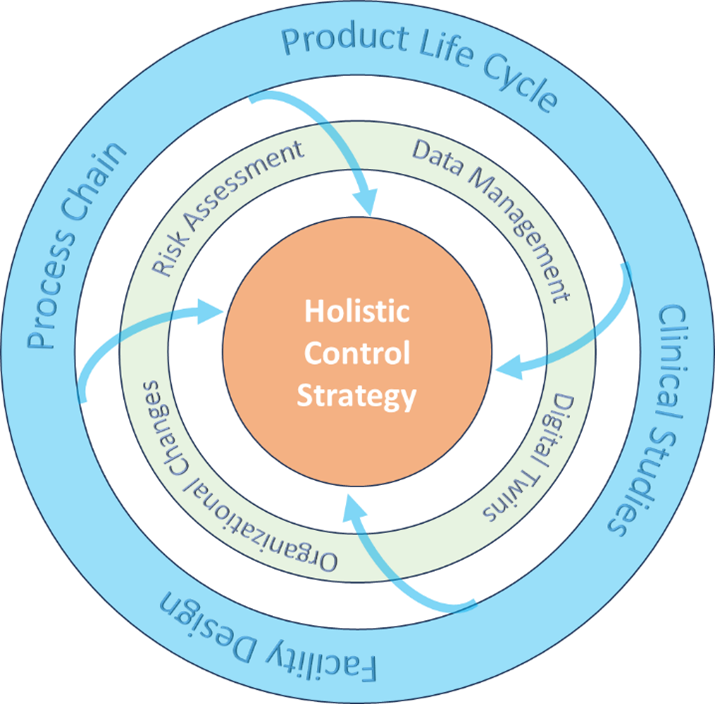
We will not repeat the gaps of the current derivation and usage of the CS . There are several disconnects inside the process chain—along the PLM, between facility design, and in clinical study design and process design. We proposed a set of tools that allow a more holistic approach by using technological enablers. We believe that this holistic CS can create immediate business benefits if the CS is comprehensive and correctly justified.
- End-to-end digital twins save more than 50% of the experiments in process characterization 21. Because of this, fewer batches are needed during development and validation when digital twins are in place. The control strategy can enable savings from accelerated tech transfer and, when in silico models are present, wider design spaces and control strategies as well. The true advantage from a holistic CS comes from broader design space and wider holistic control impact.
A holistic control strategy supports accelerated and facilitated life cycle management, and reduces regulatory CMC burden (the control strategy has fewer regulatory variations and the essential elements for product quality are correctly justified and described in the regulatory file). It also enables smoother tech transfers by including facility design limits and scale-relevant understanding in the control strategy.
With a strong control strategy, there’s more concise clinical studies and thus a higher probability of success. There are fewer design errorswhich helps avoid scale-up effects and experiencing limitations in operation. It also facilitates manufacturing authorization obtention and reduces the time for dossier approval, due to fewer quality and assurance reviews and discussions with regulatory authorities. There are reduced batch failures because of a full understanding of the process chain interactivities, quicker batch release up to real-time release solutions, and a reduced effort in postapproval changes.
A holistic control strategy supports validation and verification, as we can assure the context of use being patient safety and efficacy. Overall, holistic control strategies allow for more flexible control ranges for holistically non-CPPs, e.g., parameters that seem to be critical on one. For further insights into this topic, we encourage the reader to review the ISPE Pharma 4.0 Baseline Guide 22.