Delivering Curative Therapies: Autologous vs. Allogeneic Supply Chains

Advanced therapy medicinal products (ATMPs) are one of the most promising developments in the pharmaceutical and biotech industries in recent decades. Although there is a great promise to treat and even cure many diseases with these products, there are also unique challenges, especially with their supply chains.
ATMP supply chains face substantial challenges related to scale-up and scale-out, timely deliveries, and costs. Supply chain costs represent approximately 30% of the total cost of treatment.1 When designing efficient supply chain networks, it is critical to ensure best practices with sample tracking, package and shipping, storage solutions, software solutions, regulatory, trade compliance, customs regulations, and chain-of-custody documentation.
Cell therapy products are a subset of ATMPs. Cell-based products can be damaged due to mishandling, leading to contamination or loss of functionality. They are sensitive to temperature and stress, requiring special care and expertise in handling during transportation. These products can be classified as autologous or allogeneic depending on their starting cell origin. In this article, we explore some commonalities and differences between autologous and allogeneic supply chains.
Autologous Supply Chains
This patient-centric supply chain starts and ends with the same individual (patient). A typical autologous supply chain is shown in Figure 1. In supply chains, one of the most important metrics is lead time, which is usually defined as the time when a customer places an order to the time that order is shipped or received: from receiving patient related raw material(s) to when the customer receives the order, including converting it to a finished product, packaging, and shipping. In autologous supply chains, this lead time is also referred to as vein-to-vein or needle-to-needle time because the input material comes from the patient and the final product is administered to the same patient.
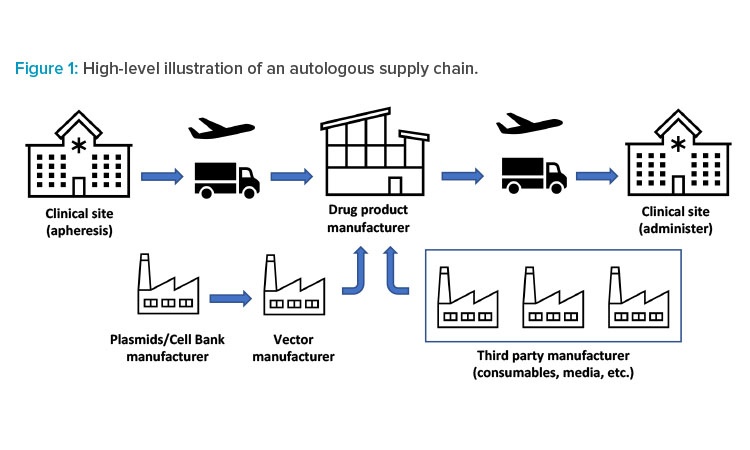
As shown in Figure 1, the autologous supply chain starts with patient material and is supplemented by other raw materials, such as vector, media, and consumables. The patient material will typically be drawn at a clinical site. However, scheduling the procedure can be equally challenging. There is a need for real-time and efficient communication among all parties involved: clinics, manufacturers, and suppliers. These interactions improve the prediction of product delivery date based on the current manufacturing capacity, which allows manufacturers to level load their resources2 and benefits patients by reducing the turnaround time.
The complexity of interaction between parties in these supply chain networks exceeds the interactions in supply chain networks of other existing industries due to the critical nature of the product being manufactured and shipped. After drawing the patient material (e.g., through apheresis), the patient material is packaged and shipped, either fresh or frozen, to the manufacturing centers. These materials are manipulated at the manufacturing site. The finished product is packaged and shipped either to a distribution center or directly to the clinical site for administration to the patient.
Allogeneic Supply Chains
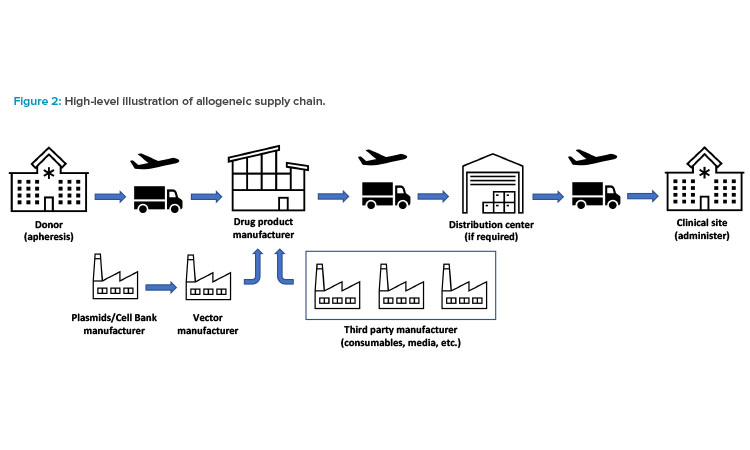
An allogeneic supply chain starts with donor cells and ends with the product being delivered to another individual (patient). A typical allogeneic supply chain is shown in Figure 2. In allogeneic supply chains, securing consistent donor material is the key to ensure manufacturing robustness. Donor cells supplemented by other raw materials, such as vector, media, and consumables, are required to start manufacturing the drug product.
The donor material will typically be drawn at a certified clinical site or a blood donation center. Handling this material is a critical part of the supply chain. It’s a complex process that includes finding appropriate donors and developing a strong donor pipeline; performing the apheresis; packaging transporting, and cryogenically shipping (as necessary) the donor material; implementing software solutions; and following regulatory, trade, and customs compliance.
Although a typical allogeneic therapy is conceived as off the shelf or make to stock, there are many other types of allogeneic therapies that may require other approaches to distribution of the drug product. The supply and distribution strategy needs to be aligned with the product and clinical needs. Universal make-to-stock therapies would allow a more traditional push model (i.e., a model where the product is placed upstream in the supply chain where it is required) for distribution in a temperature-controlled (cold) chain.
If there is a requirement for the donor to be matched in the ABO blood group system or by human leukocyte antigen (ABO/HLA) type, the manufacturer may need to maintain an inventory of multiple batches from varying ABO/HLA type donors. Maintaining inventories for these products would be more complex, but the product still would be make-to-stock. If the therapy has a very short shelf life, the drug product needs to be made to order, as in case of autologous therapies. However, as the donor material acquisition is decoupled from the patient, the turnaround time for the therapy may be reduced or manufacturing slot planning may be easier.
Key Similarities
Input Materials
Autologous and allogeneic cell therapies have similar input materials, such as cells, vectors (if cells are genetically modified), cell growth media, media additives, excipients for final formulation, and consumables. In both types of therapies, the starting cell population will need to be transformed (if cells are genetically modified) and expanded to produce the desired dose of the drug product.
Manufacturing for both therapies can start with fresh or frozen cells. Fresh cells can introduce further complexity to the manufacturing process because they need to be transported within a very limited timeframe (usually up to 72 hours, unless qualified otherwise) to the manufacturing location from the collection center. This limited timeframe can pose challenges during unexpected logistics interruptions (such as weather-related events or strikes in any part of the logistics chain).
It is important to have multiple logistics options and backup plans when using fresh cells as starting material. Starting with frozen cells is easier for manufacturing schedule because it provides the flexibility to accommodate any logistics or manufacturing delays. If the cells are frozen at the collection or apheresis center, it is important to devise clear standard operating procedures for centers to follow and to have a training plan.
In allogeneic drug product manufacturing, unless the shelf life of the product is prohibitively short, the goal is to manufacture multiple doses of the product. Thus, the scale and time frame of cell expansion, the volumes of reagents used, and the types of consumables may differ. However, the approach and management of the supply chain for the input materials (excluding cells) will remain the same. These materials can be ordered ahead of time and a certain level of strategic stock can be managed. Single-sourced raw materials are common in ATMP manufacturing and stocks should be managed accordingly.
As the number of patients going through clinical stages into commercial manufacturing increases, the supply chain may need to plan for sizable inventory, set service level agreements with existing vendors, or identify and validate a secondary source or vendor to avoid interruptions in manufacturing. The inventory will depend on patient treatment frequency, supplier lead times, and availability of alternative raw material. It is recommended to consider these alternative raw material options (if available) during initial process design or regulatory filing stages because it may be more costly and time consuming to make changes later.
Vector supply can be planned according to the patient forecast. Unlike many of the other inputs, the materials vector will be unique for each product; thus, stock management and planning for the long release times due to certain safety testing, e.g., replication competent lentivirus (RCL) is important. Supporting vector stability studies to claim as long of a shelf life as possible will make supply planning easier.
Warehouse Management
Because each batch produced in autologous therapy manufacturing is for a unique patient, the amount of raw material turnover may be faster. Though allogeneic therapy manufacturing can produce more doses, the batch sizes are much smaller than in traditional biopharmaceutical manufacturing, such as monoclonal antibodies. Subsequently, for both therapies, challenges associated with small-batch manufacturing—increased material handling and flow, inventory turns and associated cycle counts, labor hours required for pick and place activities—can be anticipated.
In both autologous and allogeneic therapies, the approach to warehouse management can follow the same principles. The raw materials received undergo an incoming quality inspection, including a visual inspection and phase-appropriate sampling and testing. The materials are held in a quarantine status until being released by the quality department. The raw material may need to be removed from the incoming packaging (wooden pallets, corrugated containers, etc.) and stored in plastic boxes or totes.
Kitting—the process of collecting parts and components per the bill of materials (BOMs) into a single kit—can be performed in advance of the production dates to simplify manufacturing operations. Appropriate kit expiration dates should be considered based on the raw materials being kitted.
Warehouses follow the appropriate engineering design factors, such as temperature and humidity monitoring and control and the proper air circulation between racks and stored materials. The layout should be designed to optimize material and personnel flows.
Packaging and Shipping
Almost all autologous and allogeneic therapies require freezing the drug product at cryogenic conditions and the use of cold supply chains to reach the patient. Packaging and handling products that require dry ice or liquid nitrogen shippers necessitate special considerations such as dedicated rooms for packing, exhaust, and monitoring. Special packaging and preparation as well as additional paperwork may be required if the product will be transported by air.
The packaging system may go through mechanical-, thermal-, or pressure-related stresses during the shipment. Thus, when designing the operational and performance qualifications, these stresses are considered and modeled carefully. The cryogenic temperatures that the packages may have to withstand could render the packaging materials brittle and may create failure points in shipments. Therefore, simulations of extreme conditions as well as actual transport conditions are covered during shipping qualifications.
It is important to include temperature monitors in the shipments; to have the proper placement of monitors; and to have the ability to start, receive, and download data from the monitors. Tamper-evident devices are also used as part of the qualification. Mock shipments for each shipping lane are recommended to troubleshoot any unforeseen circumstances.
Labeling for any products that are stored or shipped in cryogenic conditions needs to withstand these harsh conditions. Labels, as well as label ink, need to withstand a wide temperature range, e.g., from 37°C to the vapor phase of liquid nitrogen. Similar to the packaging systems, a special qualification for labels for abrasion and adhesion at the actual usage temperature, storage, and shipping conditions will prevent future deviations. For content development of labels, please refer to the widely endorsed global standards for terminology, identification, coding, and labeling of medical products of human origin, ISBT 128.3
Waste Handling
Waste management should also be given its due importance. Given the batch size of the one-per-patient philosophy, typically higher volumes of waste are generated for autologous therapies. Although the amount of waste created may be different for autologous and allogeneic cell therapies, the general principles of waste handling and management would remain the same.
Waste generated in manufacturing, packaging, quality control (QC), warehousing activities, or any other areas of the facilities are moved out in a timely manner. Appropriate staging space for waste should be allocated in the facility. Process waste, packaging waste, biowaste, etc. are handled per the company policy and local environmental policies and laws. Proper segregation of waste from drug product or raw material is achieved through physical or temporal segregation and developing and adhering to standard operating procedures.
Key Differences
Input Material: Donor vs. Patient
ATMP input materials and challenges posed by these materials are more complex compared to large molecule pharmaceuticals. Some of the input materials, such as cell growth media, media additives, or excipients for final formulation, may be similar to biopharmaceuticals. The most important input material is the donor material (in the case of allogeneic products) or the patient material (in the case of autologous products).
Donor material
Finding the right donor pool is very important. However, acquiring donor material has its own challenges due to factors such as donor eligibility, donor deferral, different motivators and barriers to donor recruitment, and decreased trust in the health care system, to name a few.4 Furthermore, even for the eligible donors, there may be variability between the donor material (though it may not be as significant an issue as experienced in the autologous therapies). As demands for these therapies increase or there is a surge in receipts of donor material, upstream cold and frozen storage requirements should be carefully scrutinized.
In addition to the challenges of finding the right donor pool, there are concerns with the donor material itself. Immune rejection can be a concern. Haploidentical matching and other human leukocyte antigen–related concerns need to be addressed. Safety, efficacy, and durability for allogeneic cell therapies is yet to be proven widely.5 However, these may become less of a concern as more therapies emerge, e.g., Ebvallo™ (tabelecleucel), which was approved as an allogeneic T-cell immunotherapy.
Furthermore, because donor material can result in a product that can go to multiple patients, the screening and testing of donor materials is tightly controlled by regulatory authorities.6 For example, apheresis blood products or mesenchymal stem cells from bone marrow are governed in Europe by the EU Blood Directive 2002/98/EC 277 and the EU Cells and Tissues Directive 2004/23/EC,8 respectively, and in the United States by 21 CFR 1271 Subpart C Donor Eligibility.9
The donor material needs to be tested for multiple communicable agents such as human immunodeficiency virus types 1 and 2, human T cell leukemia virus types 1 and 2, hepatitis B virus (HBV), hepatitis C virus (HCV), West Nile virus (WNV), and Zika virus. Furthermore, such requirements can depend on the regulatory agency’s region and jurisdiction. For example, differences exist in the expectation of the Pharmaceuticals and Medical Devices Agency (PMDA) in Japan with regard to the list of viruses of concern and testing periods compared with those given by the European Medicines Agency (EMA) and the US FDA.6
A robust donor pipeline is key for business continuity in allogeneic supply chains. The planning process should include all process steps and control points across the entire supply chain. Risk assessments should be generated and updated periodically as data becomes available.
Patient material
The most critical input material—patient cells—is a source of great variation. A patient’s age, genetic background, or state of disease can make these cells more robust or vulnerable to certain types of treatment. Sometimes the patient has gone through multiple rounds of prior line treatments such as chemotherapy and the cells can be fragile or more prone to have challenges in manufacturing. Thus, the process for ATMPs needs to be more robust to accommodate the inherent variability in the incoming patient material.
Scheduling patients has its own challenges. Scheduling is typically done manually, especially in the early stages of development. A significant amount of coordination is required between clinics and manufacturing sites with available capacities. When scaling up, adding more resources may not be economical. Effective IT systems will be important and in conjunction with the logistics systems, available data can be used to automate and forecast upcoming requests.
Variability in the arrival rate of the incoming patient material also poses a challenge for manufacturing schedules. This variability is more pronounced for fresh cells because the cells arriving at the manufacturing center have only a very limited amount of time (usually 24 hours) to be further processed. Therefore, the manufacturing center needs to always have a certain capacity available to handle this variability on arrival. This can mean overtime, 24/7 coverage, or need for additional resources for the manufacturing center.
Manufacturing: Scale-Out vs. Scale-Up
Within manufacturing, the most critical unit operation is the modification and expansion of cells. Automated and closed systems can help control and manage the process. As demand for such treatments increase, that process must be scaled as well. For autologous therapies, the process needs to be scaled out with multiple platforms and workstations, each making a single drug product for one patient (batch size of one). This may translate into the need for a larger workforce, more equipment, and more space. Whereas for allogeneic therapies, the lot size can be much larger. These therapies can be used to treat hundreds of patients. This scaling up to produce larger quantities that can be aliquoted into multiple doses to treat several patients is one of the advantages of allogeneic therapies.
The manufacturing time frame is also different for these two therapies. For autologous therapies, the vein-to-vein time (lead time) is critical because there is a life (patient) waiting at the other end. Usually, the manufacturing lead time is about two to three weeks. Uncertainties arising due to varying demands, possible sourcing issues and lead times for key raw materials, variability in the quality of patient material, arrival rate of patient material at the manufacturing center, and equipment reliability, to name a few, can influence the lead times and manufacturing costs of goods.
Rightsizing resources under these variable and uncertain conditions is very important to manage lead times and the costs of goods. Rightsizing involves estimating equipment, personnel, utilities, site logistics (material and personnel movement), spaces for production, raw materials, intermediate and finished goods staging, and support functions (e.g., warehousing, quality assurance (QA) and QC, maintenance, administration).10 Capacities should be planned to allow for surge demand when the schedules for the incoming patient material and outgoing patient therapeutics create a spike in workload.
The ATMP products are usually cryopreserved at the final stage of manufacturing and must be quarantined until all QA/QC procedures are completed and the product is released. Especially for autologous therapies, the QA/QC turnaround time becomes critical. As autologous therapies scale out, the number of samples to be tested can increase significantly. It is important to ensure that the labs do not become a bottleneck.
Because the vein-to-vein time for autologous therapies can be a few weeks, the patients must wait for a while. This makes maintenance of patient’s health or consolidation therapies important to ensure the patients can receive those cells. The time between evaluating whether the patient is eligible and the infusion can be months. That changes in the case of allogeneic therapy, where infusion can occur in a matter of days.5 With allogeneic therapies, there usually is less of a time constraint because a large batch can be produced in advance so treatment can be more readily available. However, for time-sensitive allogeneic therapies, i.e., when shelf life or hold times are shorter, their manufacturing process may look similar to autologous therapies.
Storage and Logistical Considerations
Although the storage and logistics attributes between the two therapies remain the same—the need for cold chains; packaging, shipping, and labeling considerations; sample storage; distribution challenges; chain of custody; strategies that include processes and procedures for the return and destruction of product, in compliance with the FDA, EMA, and other regulators—their scales are different. A few key differences are further explained next.
Cryopreservation is an important factor to be considered before and during shipping final products for both therapy types. Because autologous therapies cater to the same patient (have a batch size of one), the number of doses required to be frozen before shipment is not large. On the other hand, allogeneic therapies require larger storage capacity because one batch can be worth thousands of doses for multiple patients rather than a single dose per patient. Cryopreservation of large number of doses requires specialized equipment. Also, it is important to decide if the cryopreservation can happen in the same suite or adjacent suites.
The number of samples that need to be stored is higher for autologous therapies, including starting material from the patients, the final drug product, and everything in between. This may necessitate larger storage space or different storage strategies. In autologous therapies, the patient and their location are known from the outset. Although managing this cold chain is challenging, the distribution can be more complex for allogeneic therapies. The latter can be distributed to patients across a wide geographic area. This presents a unique challenge for the allogeneic therapies: identifying where to position product inventory, inventory quantities at these locations, and distribution channels for delivery to an undefined network of caregivers, all while maintaining product quality and service level agreements.
Universal make to stock is typically what people think of for allogeneic therapies and would allow a more traditional push model for distribution under a cold chain. A hospital orders, or has a standing order for, a therapy and the hospital may maintain a stock, like most other medicines. In the made-to-order approach, a pull system is employed, which requires coordination with the manufacturer’s production slots. This model is usually adopted for autologous therapies. However, the front end is decoupled from the patient so an inventory of work in progress is typically maintained to reduce time from order to delivery. The made-to-order strategy may be applicable for allogeneic therapies with a very short shelf life or in the case of a large number of donor matched types (e.g., multiple-point HLA match).
To ensure patient and product safety and reduce human errors, a well-designed tracking system covering chain of custody and chain of identity is necessary. When developing or selecting the tracking system, ensure it can accomplish all needed tasks, such as linking geographic tracking data, monitoring temperature, and tracking and documenting chain-of-custody data in real time. Traceability may not be as critical in allogeneic supply chains as it is in the autologous supply chains. Orchestration plays a significant role in the autologous supply chains. However, chain of custody is important in both cases to ensure patient safety and product quality. Collaborating with relevant players in this chain of custody to share information can provide the flexibility to balance fluctuations in supply and demand.
| Attributes | Autologous | Allogeneic | Points to Consider |
|---|---|---|---|
| Starting cell material | Patient | Donor | Allogeneic: Business continuity and securing donors |
| Chain of custody | Yes | Yes | |
| Chain of identity | Yes | Yes | Autologous: More critical for this therapy |
| Manufacturing expansion | Scale-out | Scale-up | Autologous: Facility design implications |
| Sample storage space need |
Higher | Lower | Autologous: Facility design implications |
| Availability of treatment | Patient waits | Potential shorter patient wait time |
Autologous: Maintenance of patient’s health or consolidation therapies |
| Supply chain model | Pull (made to order) | Push (make to stock) | Allogeneic: If the therapy has a short shelf life, it may need to be made to order |
| Drug product inventory | Not available (unless multiple doses can be manufactured at once and can be readministered) |
Available | Allogeneic: Evaluation of drug product inventory quantity and locations |
The individualized nature of cell and gene therapy products also requires meticulous tracking of the patient material and final products. Maintaining identity and custody at each step of the supply chain is critical to ensure patient safety. Each therapy product should be tracked to the patient and the chain of custody should be visible to coordinators. Investing and adopting the right level of automation strategy is important to achieve this capability.
Conclusion
The curative promise of ATMP products makes them highly attractive solutions for certain diseases. However, managing supply chains where the patient or donor is a critical part of the supply chain can be challenging. Key similarities and differences have been described at a high level (see Table 1), but other aspects should also be considered when designing a robust supply chain.
Several steps in the supply chain are vulnerable to external issues like weather-related disruptions, macroeconomics, global political scenario, and raw material availability. Thus, supply chain mapping, exceptions planning, what-if analysis, corrective actions, and escalation protocols become important. These can help improve supplier relationships, manage overall costs, and, most importantly, save lives.
The level of automation and digitalization in manufacturing processes and supply chains should also be explored. An economically feasible and regulatory compliant digital roadmap that allows for tracking, collecting, and sharing or reporting the right data should be developed. The data-tracking equipment and protocols that suffice in the ATMP research lab may not be transferable to a commercial-scale, GMP-grade aseptic manufacturing environment. An ATMP facility might receive and release thousands of product batches over the course of a year. These facilities, and their supply chains, will need robust strategies to prevent product mix-ups and ensure the integrity and traceability of every one of those batches, including samples sent to the QC labs.
Clinics and hospitals are also an integral and crucial part of the ATMP supply chains. It is important to ensure trained staff is available at these sites for apheresis, as well as when the product arrives and is ready for administration. Appropriate equipment and processes for thawing, washing (if required), and administering should be available at these sites.
The complexity of the ATMP supply chains is unique. These supply chains involve clinics, transportation partners, third-party material providers, manufacturers, and biopharmaceutical and biotechnology companies. Partnerships for all supply chain players are also complex: There are scheduling and logistical challenges; issues controlled by contracts and confidentiality agreements, training programs, and significant amounts of documentation that often are specific to individual hospitals. The supply chain challenges are considerable, and it is never too early to start addressing them, especially when the goal is to deliver the curative power of these therapies to the patients at the end of these supply chains.
About the Authors




