Operational Risk Management in Global Supply Scenarios
The current regulatory framework in the pharmaceutical industry places pressure on marketing authorization holders (MAHs) to demonstrate quality oversight, and a systematic risk management process is a prerequisite for avoiding compliance and productivity pitfalls. This article focuses on options to improve day to- day operations and to ease decision-making by integrating operational risk management into a company’s quality system.
Key risk management concepts were introduced to the pharmaceutical industry about 20 years ago when, for example, the World Health Organization recommended that the food industry’s hazard analysis and critical control point (HACCP) methodology also be applied to pharmaceuticals with a focus on monitoring critical control points (CCP).1 Various industry stakeholders and regulatory institutions now seem to issue new risk management guidelines nearly every year. In particular, the US FDA initiative for cGMPs in the 21st century2 and ICH Q93 triggered a broader discussion of risk management requirements.
Risk management is mentioned in many ICH guidelines (specifically, ICH Q84 and ICH Q105) and is part of the EU GMP regulations.6 ICH E6 (R2)7 situates risk management in the Good Clinical Practice (GCP) scope. In the medical devices industry, both ISO 13485, which concerns the quality management system8 and ISO 14971, which addresses risk management,9 are required for market approvals.
Risk management is also an important element of the ISO 9000 series, which addresses quality management.10 ISO 31000 has a broader focus on risks in organizations. 11 This standard may influence future directions in the pharma industry as it applies to the whole organization, rather than just quality.
Despite this range of regulations, the pharma industry has not made much progress implementing risk management strategies in recent years. The methodologies listed in Annex 1 of ICH Q9 can be widely applied. 3, 12 However, a single approach—failure mode effects and criticality analysis (FMECA) in combination with spreadsheet applications—predominates. As Greene and coauthors have stated, the value of quality risk management in the industry “has not yet been realized”.13 Moreover, there are reports of companies applying risk management in situations where it is inadequate or inappropriate—for example, to justify noncompliance situations or as a proxy for proper root-cause investigations. This results in the industry facing in-creasing risks rather than getting existing ones under control.14
These observations align with perceptions that the pharma industry has made less progress toward continuous improvement and innovation compared to other industries.15 In sum, although risk management tools are applied here and there in daily quality operations, a systematic approach to risk management is missing and there is room to improve.
Global Supply Chain Challenges
The pharmaceutical industry underwent remarkable changes in the 1990s. Mergers and acquisitions, new launches of blockbuster drugs, the growing number of older adults, and a high demand for drugs to manage hypertension, cancer, and diabetes indicated a bright future for pharma. In this era, supply chains were fully integrated from API synthesis to finished product market delivery.
Later, the supply chains became more and more diverse and complex as certain drug patents expired. Generic manufacturing, first in the US and Europe and later in Asia, together with price pressures from buyers, led to more risks.16, 17, 18, 19
Though every step in a supply scenario can be performed by someone else, the MAH remains in charge. In the view of regulators, the MAH cannot delegate the responsibility for the products it markets.
The consequences of this requirement are not always clear. MAHs are often just sales organizations without the resources to oversee all the activities along the value chain. GDP regulations brought a bit more clarity about what is required, but these regulations were not definitive. European inspectors have recognized a lack of quality oversight, and the EMA issued a reflection paper20 on the topic for public comments in January 2020. It is obvious that processes must be developed for MAHs to gain complete oversight. These processes must be integrated into the quality management system. In addition, the MAH is also responsible for pharmacovigilance. Small organizations often neglect these requirements and outsource pharmacovigilance services, which again diminishes the MAH’s oversight. Finally, regulators expect the MAH to maintain its ability to supply its products. Risk management is probably the only concept that can address risks impacting quality, patient safety, and deliverability. The whole organization must be involved to reduce and manage risks. Risk management ultimately concerns the freedom to operate.
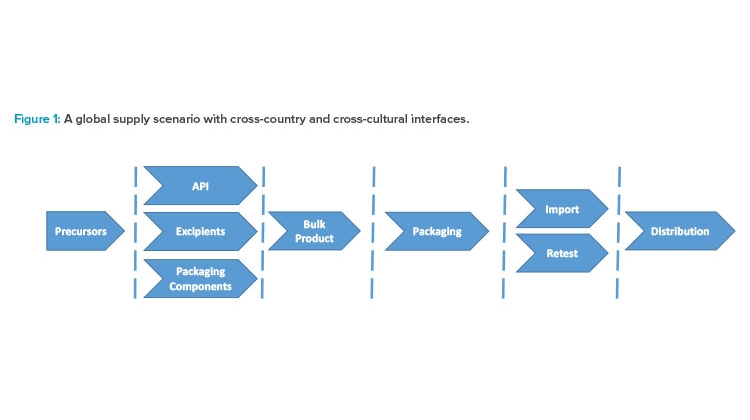
Figure 1 illustrates the complexity of a supply scenario with manufacturing in Asia and, export to Europe and the US, with retesting, batch certification, and final distribution. Each group of chevrons belongs to a different site and country. A dashed line indicates an interface where errors may occur and miscommunication may happen. If errors slip through, they can adversely affect patients. Unavailability of drugs due to challenges in foreign countries is a serious problem in Europe, particularly during the COVID-19 crisis. The tendency to “insource” (i.e., bring in house) services that were previously outsourced is now evident in the European sector, despite the huge effort this requires for technical transfers and the expenses of re-registration fees. New risks will likely emerge if competencies and industry knowledge need to be reestablished in Europe as part of this trend.
Some quality professionals believe that MAH oversight can be achieved by service-level and quality agreements.21 The expectation is that everything will run smoothly once these documents are in place with shared responsibilities defined. Practice, however, shows that communication between contract partners tends to cease shortly after paperwork is done. At this point, risk management strategies are critical.
To gain oversight, the MAH must know what can go wrong. Long before the specific risks of the manufacturing process are addressed, it is important to understand the interfaces. The dashed lines in Figure 1 represent points of control, but they also represent points where the flow of goods and data can become blocked, or information can get lost. People in charge must establish and maintain their communication channels. This ensures a state of control and initiates continual improvement.
Products are endangered by temperature, humidity, and mechanical influence. These risks are nowadays quite well under control. However, successful batch release decisions depend on reliable document exchanges. Unfortunately, the required documents may be missing, wrong, or unclear. Documentation errors are early warning indicators or proxies for underlying weak points in the organization of work. When data transfers from one IT system into another occur in conjunction with miscommunication and negligence, the results may include data integrity problems, human errors, and wrong decisions; ultimately, the MAH may lose the necessary control of situations and become incapable of delivering products.
Furthermore, complex transportation routes may also be at increased risk for fraudulent activities. The answer to this is serialization. Modern enterprise resource planning systems allow companies to collect the requisite data. Managing risks involves detection of anomalies, from end to end, in data transfer processes.
The division of labor, price pressures, and a lack of communication between the individuals have led to serious violations of GMPs. The high number of US FDA warning letters issued to manufacturers in China and India in recent years is a symptom of serious inherent risks in global supply chains22 In some cases, European MAHs have not received important information promptly and have been surprised by emerging problems. Recalls are required if an unsafe product is already on the market.
Risk management focuses on risk prevention. Pitfalls can be avoided when the appropriate controls are in place. These controls must cover the entire product life cycle.
In manufacturing, risk management must focus on technology and process robustness. Solid oral dosage forms are the largest category of pharmaceutical products. Nevertheless, progress in risk management in this field came late. Risk management and quality by design (QbD) are the essential elements in the quality system, which is the right approach to develop scale-up processes and increase robustness. Good indicators for process robustness are content uniformity of blends, reliable particle size distributions of granulated material, and reproducible dissolution rates. Control charts clearly show any adverse trends and illustrate whether the manufacturing process is robust enough (i.e., is under control).
Annual product quality reviews help stakeholders analyze the centricity of a process between the lower and upper specification limits. Data from these reviews can be used to calculate the process capability index. Descriptive statistics are a central element in risk management. It is in the interest of the MAH to verify that contract manufacturers have the requisite competencies to react early to trends and to continuously improve processes that ensure robustness.
Another area for risk management is cross contamination. For APIs, key risk factors for cross contamination can be attributed to synthesis routes; use of recycled solvents and catalysts; material storage and flow in factories; and multipurpose equipment, transfer tubes, and containers, and their cleaning operations.
HVAC design has shown to be troublesome in production facilities. The accumulation of dust and dried-out coating residues in parts of the equipment and in filters, together with pressure variations, may lead to the release of contaminants into fresh blends. Traces of other chemicals may not be detected by routine testing. It is easy to claim that cleaning validation prevents this problem. However, warning letters show that these risks are not always under control. This must be addressed in the planning phase, and risk management is the tool of choice.
Process robustness, predictive maintenance, and effective barriers to prevent cross contamination are assets at every manufacturing site.
To manage risk at the interfaces, further measures are required up front. Risk management and quality planning must include training of employees and communication management in global supply scenarios before adverse trends lead to nonconformities. A “person in the plant” strategy (i.e., an employee of A works at site B, and vice versa) is one promising approach to manage such risks.
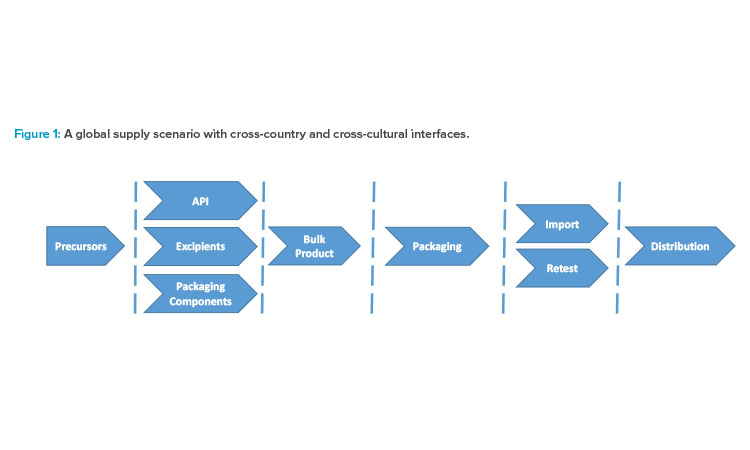
For countries with nonharmonized GMP regulations (countries without mutual recognition agreements), a retest of imported material is required. If both the process and the testing methods are robust, a loss of control it is extremely unlikely (see Figure 2).
To recap, risk management must trigger process robustness, prevent cross contamination, and ensure communication crossing interfaces. With these key factors in mind, it becomes obvious that risk management embedded into the quality system can offer a lot of advantages.
Organizing Operational Risk Management
Organizing operational risk management into modules and phases can be an effective approach. Figure 1 may be useful to define modules/work packages to implement risk management across all operations. It is advantageous for the MHA to create a risk assessment package for each chevron and its input and output factors shown in the figure.
Asking “What is the risk here, and how do you want to manage it?” is the starting point in day-to-day operations and decision-making of a mature organization.
Following ISO 31000 [11], the whole organization is involved in managing risk. The organization’s top management is in the driver’s seat, and the project starts with assigning the responsibility on a high level. No management commitment means no progress. Asking ”What is the risk here, and how do you want to manage it?” is the starting point in day-to-day operations and decision-making of a mature organization.
To set up and maintain a risk management system, the organization will require a few skilled risk management ambassadors for training, support, and internal communication. These ambassadors must spread the seed for a company’s risk management ambitions. As discussed by Gigerenzer,22 increasing the risk savviness of individuals and, finally, the whole organization takes effort.
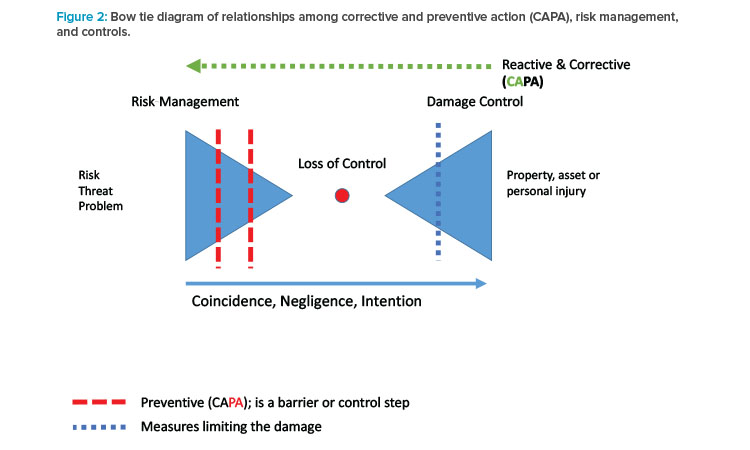
Assuming that management is committed, and the ambassadors have some credibility in the organization, a risk-based system to manage operational quality and supply risks can be achieved in three phases (Figure 3).
The first phase provides the foundation for the system. Its outcome is an operational risk management report, which is the basis for the risk register or risk library. This phase is described in greater detail in the next section (Risk Identification).
In the second phase, selected remediation activities can be initiated to directly address specific risks identified in phase 1.
For example, if the report generated in phase 1 shows many problems with documentation, a corrective and preventive action (CAPA) project could be triggered to change instructions that are unclear, too long, error inducing, or otherwise problematic. Improved (modular) templates designed to help operators follow a process can reduce errors in daily operations.
In another example, CAPA might address missing data in handwritten batch records, erroneous yield calculations, and missing controls. One option might be to make the right place to document data more obvious. This basic change may lead to fewer documentation errors and less need for retrain-ing. “Nudging” is a current buzzword in this context.
When teams work together, data control and data integrity (i.e., adherence to ALCOA+ principles) are put at risk by sharing, storing, and retrieving documents on shared drives. Changes such as establishing a directory plan defining a process-oriented filing system or using a cloud-based document management system may help reduce these risks.
All such phase 2 remediation projects must be appropriately staffed and executed. With a few projects like those described here, the organization harvests low-hanging fruits. Employees and contractors will see that there is something in these efforts for them and will likely accommodate new ways of working. However, individual CAPA measures typically deal only with single deficiencies and are therefore inadequate to fully manage risk.
In the third (routine) phase, the organization uses risk management in daily operations. When new risks are discovered, they are routinely processed and remediation actions are endorsed by an operational quality (risk) review board or similar entity. As noted previously, the risk ambassadors must look beyond single events and ask what is wrong with the system.
In this phase, continual improvement, risk management, and quality review come together, and the organization can control quality issues quickly and effectively. A defined improvement project is much easier to handle than checking long CAPA lists and wasting time with “number of CAPA overdue” key performance indicator tracking.
Risk Identification
To implement operational risk management, it is important to get the big picture first. Risk management depends on fast and complete knowledge of potential risks. This is related to the concept of availability. Even if a risk is known, it may not be present and documented in a certain situation. It might be identified and documented later, but in a completely different context. It is therefore important to follow a systematic approach to identify risk (i.e., potential nonconformities). Information sources include:
- Electronic common technical document (eCTD) Modules 1 and 323
- Commercial contracts and technical agreements
- Contact partners (regulatory support, logistic services, laboratory operations, etc.)
It is also important for the risk management team to get in touch with other stakeholders and employees directly involved in related manufacturing and supply operations to determine what they know about risks. For example, shop floor operators may have tacit and undocumented knowledge. They know the details and can contribute to risk identification and management.
For each operational module (Figure 1), the risk management team should start by collecting as much data as possible. This involves:
- Studying the module in the registration dossier
- Looking into master batch records and recent product quality reviews
- Consulting audit reports
- Calling people identified in the contracts and asking questions like:
- What happened previously?
- What kind of deviations are observed?
- Which changes have been made in the process and why?
- What risk management efforts have already been implemented?
- What kind of risks are perceived?
- Are there hidden flaws or weak points in the process?
Risk managers should use keywords like those used in the hazard and operability study (HAZOP) methodology. They should also think in scenarios and ask, “What happens if…?” or “What can go wrong here?” It is always better to overestimate a risk than to be surprised later.
It is also helpful to query for public information, such as warning letters or reports published by competent authorities, and raise the question, “Can similar things happen in this unit operation as well?”
Controls and checks must be identified, agreed upon, implemented, and communicated. Quality oversight and communication through the interfaces are essential. Open communication on trends and near misses must be shared. Regular contacts between the risk management team and the people in charge are crucial. The risk management ambassador (or a subject matter expert in relevant unit operations) therefore plays a key role in facilitating risk identification.
The next step is to put together a list of the information received—the risk register or risk library. In the modular approach, the lists established for each chevron shown in Figure 1 will not be long—maybe 30–50 line items that are shared among similar unit operations.
The risk management team may hold brainstorming sessions to narrow down the line items by looking for common root causes, synonymous expressions, and redundancies in the issues they have collected. If the risk managers feel something is missing or contrary to their own experience, they may arrange a second round of interviews or closely examine specific issues discovered.
It is not required to collect and document complete investigation reports. Instead, the risk management team can move one level up and investigate root-cause summaries, or judge intuitively what the problem might be. Auditors typically have a good sense for operational flaws. Simply asking them can yield good information.
The final step in collecting data is to identify appropriate categories. Use a classification that suits the organization’s needs, such as 8M (machine, management, material, measurement, men, methods, mindset, mother nature) or 5P (people/personnel, processes, product, performance). to prioritize where the quality system needs to be improved.
Accurate classification of documentation errors is crucial. Useful designations include missing, wrong, not clear, misplaced, and lost. All aspects of a document’s life cycle must be addressed.
Categorizations help establish a structure, but issues can sometimes be categorized in multiple ways. The key is to be comprehensive and to clarify how categories are defined. For example, “management” could mean the style of direction or might be focused on budgeting or staffing. “Process” might be defined as “dealing with performance issues, such as drifting or unstable performance, with too many deviations.”
Another way to classify risks is by their impact. Risks may lead to personal injuries (e.g., hospitalization of a patient); damage to assets (e.g., loss of a customer or buyer); property damage (e.g., material loss due to a quality defect); GxP violations and inspection challenges; and lost productivity.
Risk Evaluation
For FMECA, which is the most frequent method of analysis used in pharmaceutical risk management, risks are described in the three dimensions: impact, likelihood, and detectability. Words like low, medium, and high, or minor, major, and critical, are used to rank risks by each dimension on an ordinal scale.
The risk manager should make a choice about how to rank risks but should not lose sight of the bigger picture.
A practical hint is to omit the valuation of likelihood from the first rankings. It is difficult to assign a likelihood to a single event, and it is not easy to identify the number of conforming events that will be appropriate to use for later rankings. It is easier to think initially about risks in terms of prevention and control measures.
With the dimensions of impact and detectability in mind, the risk manager can group and prioritize risks. If there are many risks, a simple (3 x 3) matrix can be used to cluster them. Risks with a higher rank cluster on the upper right, and the ones with lower values on the lower left. Different diagrams can be established—for example, there might be a diagram for each unit operation, diagrams for the interfaces being considered, or one diagram for each impact category.
The clusters may show different patterns, and pattern recognition can be advanced through artificial intelligence. Mighty tools such as multivariate statistics, cluster analysis, principle component analysis, and data mining are infrequently used, but they can help stakeholders make better decisions.
Risk Control
In phase 3 of risk management, barriers or controls can have a broad meaning. Detectability of an error mode is not always related to sensors and technologies. Organizational measures are equally important, so that coincidence, negligence, or intention does not lead to a loss of control (see Figure 2).
To maintain momentum in operational risk management, a quality risk review board brings risk assessment, review, and communication together. The board can also facilitate the use of new risk management tools and training for risk managers in their use. The organization will recognize their benefits as soon as the first effects become visible.
After implementation of operational risk management, regular ad hoc reviews should take place to ensure that improvement projects are on track. The role of the risk manager is to manage the risk portfolio and to prepare decision-making by the risk management committee or team. Applying a plan-do-check-act cycle ensures a constant stream of information, acknowledgment, and risk mitigation actions is maintained.
Conclusion
A risk management system can be embedded into a pharmaceutical quality system by first identifying the elements in a complex supply chain. Unit operations and interfaces are the primary work packages to be considered. Risk assessments on a unit operation level result in a risk register or risk library, which is maintained by risk ambassadors. A single process for risk management embedded in the quality system integrates project controls, management, and product quality reviews. Modern technology can be used to present the right metrics, allowing the organization to make timely, fact-based decisions about operational risks.
About the Author



