Post-Approval Change Management for Cell and Gene Therapy Products

Cell and gene therapy (C>) products represent a significant step forward in patient treatment and often offer unique patient benefits. However, product developers face significant hurdles within the regulatory landscape. The tools laid out in the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Q12 guideline: “Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management”
Regulatory Challenges for C> Product Developers
Developers of C> products face significant complexity within the regulatory framework. This field is relatively new and less established than other traditional biotechnology fields. Therefore, both development and regulatory frameworks are evolving continuously in line with emerging information. This leads to a complex and uncertain regulatory landscape with significant hurdles. Lack of international harmonization across regulatory agencies on approaches to these products adds to the challenges, particularly in the post-approval space. For future development of these products, speed and agility should be considered in the context of this framework.
One possible solution to these challenges is the Post-Approval Change Management Protocol (PACMP) concept, as outlined in the ICH Q12 guideline. This concept could be used in novel ways and leveraged to facilitate the updating of certain product quality attributes, such as product-related impurities specifications, based upon evolving understanding and increased clinical experience and product manufacture. This concept is outlined in this article.
ICH Q12 and the PACMP
The ICH started work on a specific guideline to address the issue of life cycle management from a science- and risk-based approach in 2014. This resulted in the ICH Q12 guideline, which reached Step 4 in 2019 and has been in ongoing implementation globally since this time. ICH Q12 sets out to provide a framework for the management of post-approval change and is designed to increase the predictability and transparency of change proposals, both for the license holder and the competent authority.
ICH Q12 introduces several tools for the management of regulatory change. Of particular interest to this discussion is the PACMP. Tools like the PACMP have been available in some jurisdictions for many years, so technically this is not an innovation.
Traditional Post-Approval Change
In a traditional post-approval change interaction, the applicant makes one submission to the agency in which the scope of the change is outlined, including an outline of experimental and confirmatory work necessary to support the change and the data from this work. For a change to a well-characterized biotechnology product, this would entail a description of the change, a risk assessment of the impact of the change, a presentation of the comparability activities and data (as envisaged in ICH Q5E), and data from any stability studies considered necessary.
There are two major issues with this approach. First, logistically, the applicant must wait until all the data is gathered before making a submission to an agency. Given the potential requirement for stability data, waiting for this data can significantly lengthen the process. Second, the agency reviews both the applicant’s strategy for the change and the resulting data corresponding to that strategy at the same time.
Consequently, if the agency has issues with the path the applicant has taken and believes that additional or different experiments are necessary to support the change, this could jeopardize the approvability of the change and lead to significant delays in implementation. This can be especially trou-blesome in situations where the change is complex or where there is limited guidance.
Post-Approval Change Under the PACMP
With the PACMP, the applicant makes two submissions: one outlining the scope and strategy for evaluation of the change and the supporting work to be performed and a second subsequent submission providing the resulting data from the agreed-upon supporting work. Although this may seem to be dou-ble work, this approach offers transparency, predictability, and efficiency over the traditional approach.
First, the submission of the strategy is freed from the need to wait for generation of all the data. Thus, this can be done at any time once the strategy for the change and the required supporting information has been established. Not having to wait for the data constitutes a significant time savings.2 Second, provided that the data meets the agreed-upon protocol, the subsequent implementing submission is expected to be a lower category of change, usually moving from prior approval category to a notification, providing a significant efficiency once the appropriate results are available.
Finally, and perhaps most important, the applicant and the agency have a chance to discuss and agree on an appropriate package of information to support the change before any significant work has been initiated. Crucially, this agreement is binding, coming as it does from the approval of a regulatory submission. The probability of any agency queries sufficiently major enough to derail the progress of the change is, therefore, markedly reduced so long as the agreed-upon strategy and data package are delivered.
In addition, ICH Q12 makes it clear that the PACMP need not be confined to one change only. Written well and with the appropriate justification, a PACMP has the potential to cover similar changes multiple times for a single product or similar changes across multiple products. PACMPs are increasingly seen as a useful tool by many agencies and almost as a first step into implementing ICH Q12 globally.
Several pilot programs have included the use of PACMPs, and there is increasing interest in PACMPs across many agencies. Indeed, the use of PACMPs as a transparent and efficient regulatory instrument has been reflected in the European Medical Agency’s PRIME toolbox 3 as a useful way to manage the kinds of complex changes envisaged in accelerated development scenarios.
Use of PACMPs offers a structured and systematic approach to managing changes in product quality attributes, and specifications, over time.
Opportunities with PACMPS
The ICH Q5E guideline “Comparability of Biotechnological/Biological Products” 4 is widely regarded as a reliable framework for assessing comparability in biotechnological and biological products, such as monoclonal antibody therapies. This framework should be applied to C> products, using a risk-based approach that considers their unique characteristics.
Due to the innovative nature of C>s, flexibility is necessary to maintain high-quality standards because traditional methods of demonstrating comparability may not always be appropriate. It is crucial to adapt and tailor the assessment process to effectively address the specific challenges associated with C> products, thus ensuring their exceptional standards are upheld.
Use of PACMPs offers a structured and systematic approach to managing changes in product quality attributes, and specifications, over time. This proactive strategy not only allows for adjustments driven by advancements in manufacturing knowledge but also enhances product adaptability to meet evolving regulatory requirements and patient needs. In this article, we propose transformational use of PACMPs for effective management of specification changes for C> products.
Specifications
Current state manufacturing processes for autologous chimeric antigen receptor (CAR) T cell products often result in a large degree of nonconforming results. This is due to the inherent variability in the cellular starting material for the process (i.e., patient material including those with significant illness). Initial specifications of these products are often based on limited clinical experience, resulting in the setting of limits that are frequently not reflective of the process capability.
If an out-of-specification (OOS) result is observed for a patient lot, a quality event is initiated with laboratory and manufacturing process elements investigated. Each batch that generates an OOS result has an individual impact assessment performed. In addition to the impact assessment, batches undergo review through relevant safety and quality committees to assess the manufacturing/testing process in conjunction with the medical status of the patient to determine a recommended action plan for batch disposition determination.
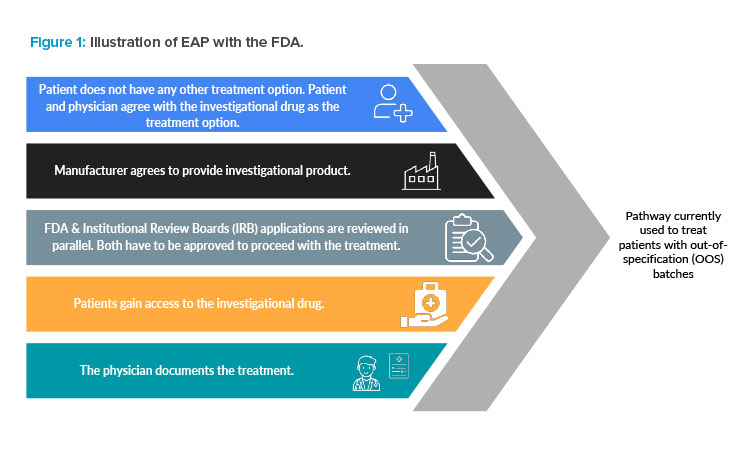
At this point, if the product poses a safety concern to the patient, then the batch is not administered. However, if there is a high degree of confidence that the benefit-risk balance favors continuing with the batch, oftentimes a physician is contacted, and options are reviewed for their decision on acceptability of the material. In the event a physician chooses to still administer the nonconforming product, exceptional release of these batches is often pursued through a single patient clinical protocol (single subject investigational new drug [sIND]) or a managed access or expanded access program (MAP/EAP) of the manufacturer. Such pathways are outlined in 21 CFR 312 Subpart I for “Expanded Access to Investigational Drugs for Treatment Use.”5
The EAP in the US, summarized in Figure 1, “is a potential pathway for a patient with a serious or immediately life-threatening disease or condition to gain access to an investigational medical product (drug, biologic, or medical device) for treatment outside of clinical trials when no comparable or satisfactory alternative therapy options are available.”6 Many products in this space are only approved for later lines of treatment, resulting in patient populations that fit this description.
Products that fail the commercial specification can often meet the envelope of specifications used in the clinical investigations, which were the basis for regulatory approval. Additionally, clinical specifications are often established earlier in the product development life cycle and therefore are broader.
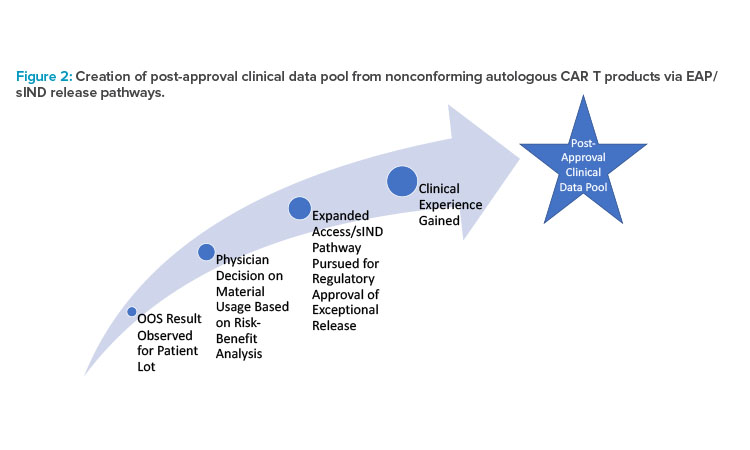
As a result, significant effort by companies is often put into the EAP. Through this process, alignment with health care providers (HCPs) and regulators on patient suitability is reconciled with quality/GMP release considerations as quickly as possible to serve the patients in need. Over the course of the product’s life cycle, significant data is accumulated for lots outside of the commercial specification. As such, an opportunity presents itself to explore utilization of this data set, where clinically relevant data is accumulated as a direct consequence of continuous process performance and patient need (see Figure 2).
Additionally, the goal of the third stage of process validation is “continual assurance that the process remains in a state of control (the validated state) during commercial manufacture.”7 An ongoing program to collect and analyze product and process data that relate to product quality must be established.8
In the instances where exceptional release is pursued for nonconforming CAR T products, a manufacturer can leverage clinically relevant data outside of commercial specification to ultimately expand the specification range of that attribute while demonstrating control. A nonclinical protocol tracking the safety and efficacy performance of these OOS lots can capture this data. This will build the necessary rationale to produce a statistically and/or clini-cally relevant specification range more reflective of the process capability while demonstrating no patient impact.
A PACMP could be an appropriate way to negotiate changes to specifications leveraging the large and evolving post-approval data gathered on these products. A PACMP could illustrate to an agency how a company intends to use this data to modify the specification. This could be negotiated between the applicant and the agency, and a suitable path forward agreed upon, including the potential to downgrade the implementing submission.
Elements used for specification changes
Having clinically relevant specifications reflective of the true process performance also allows manufacturers to focus resources on more impactful investigations pertaining to true outliers from the process, not marginal OOS events from commercial specifications based on limited data that ultimately have no impact on patient safety and efficacy.
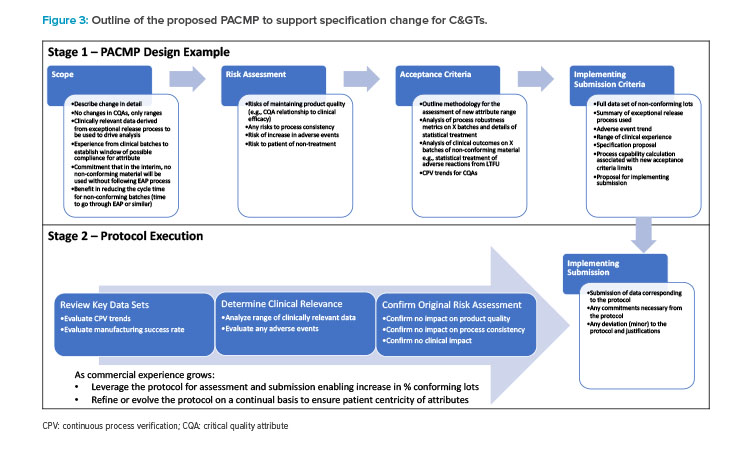
A conceptual outline of a PACMP for this kind of specification change could pull this information together to support a change, as illustrated in Figure 3.
Stage 1 is the design and submission of the protocol proposed for the change. This protocol is structured as follows:
- Outline the scope of the change in detail. (For example, the scope of the change is limited to the acceptance limit or range for the specification. Critical quality attributes and the analytical methods used for the attribute will not be changed.)
- Discuss the potential risks in making this change and how these can be mitigated, taking account of the risks and benefits to the patient.
- Outline the criteria that will be used in the assessment of the change.
- Propose the contents of the implementing submission and the reporting category.
Stage 2 is the gathering, review, and assessment of the data aligned to the protocol and the subsequent submission of this data to a regulatory agency.
As previously stated, assessing comparability for C>s is not simple and can require a combination of both analytical data and some confirmatory data from clinical use. In the PACMP, it would be anticipated that the applicant would illustrate how a justifiable number of batches could be assessed to establish an appropriate range of acceptability. This range of acceptability may be related to either the clinical materials or to a range of OOS material, shown during long term follow-up (LTFU) activities to present low probability of clinical risk to patients. All of this would be outlined with appropriate justification in the PACMP, which is subject to regulatory approval.
Upon accumulation of sufficient data, as agreed on in the PACMP, a specification change (traditionally filed as a prior approval supplement [PAS]) can be pursued as a lower reporting category (i.e., CBE-30). Furthermore, over the course of the product life cycle, as additional data is accumulated as part of the post-approval clinical data pool, additional CBE-30s can be submitted, modifying specifications and enabling a cycle of continuous improvement and understanding of the product.
Given the frequency of nonconforming commercial products in traditional autologous therapies, a future state quality system inclusive of a manufac-turing outcomes-based approach is desirable to minimize the need for EAP or sIND avenues. By having clear pathways to establish patient-centric specifications, the burden on sites, HCPs, and patients can be reduced and treatment options can be accelerated.
Furthermore, significant advantages exist pertaining to simplifying the notification process of release for OOS products. By linking this process simplification with a cycle of data collection/specification refinement, great benefit to patients through expanding access to these innovative, lifesaving treatments can be achieved. Future autologous products should consider defining such quality strategies proactively as part of their quality management system (potentially through PACMPs), leveraging risk management principles. Key questions to consider as part of this strategy development include:
- What is the appropriate nonclinical tracking mechanism for patients receiving nonconforming products during LTFU?
- How many data points are needed to justify expansion of commercial specifications?
- The goal of the third stage of process validation is continual assurance that the process remains in a state of control (the validated state) during commercial manufacture. A holistic picture of the manufacturing process and its performance relative to the specifications is needed to justify further modification. What models and/or principles can be applied to further bolster the argument of modifying specifications while maintaining control?
- By implementing a cycle of continuous improvement based on the increasing clinical data pool accrued by nonconformances, how are process changes and improvements being factored in to reduce and ultimately eliminate the need for such a cycle?
Life Cycle Management
The rapid advancement of scientific understanding within the pharmaceutical sector has paved the way for ground-breaking modalities like C>s and personalized medicines. However, this surge in innovation is placing increasing strain on the current process of justifying specifications between health authorities and applicants.
The rapid pace of scientific advancements necessitates an alternative approach to effectively navigate the evolving landscape of pharmaceutical development and regulatory processes. Typically, numerous C>s are approved for the market with a limited data set and on a small scale. Consequently, changes are bound to occur as more data is gathered about the product, as the manufacturing process is improved, and as the scale of production expands.
The scientific knowledge that the pharmaceutical industry is gaining in the field of C>s opens possibilities for increased collaboration between industry and regulators. This collaboration can result in the formulation of specifications that prioritize the quality, safety, and effectiveness of new medicines, ultimately meeting the needs of extremely ill patients.
Additionally, it can play a vital role in establishing resilient supply chains, enhancing the accessibility of these medicines for patients in need. Due to the limited number of clinical lots and process characterization data available during the submission stage of C>s, regulators and industry may collaboratively set interim commercial specifications.
These specifications are determined based on the potential risks to patients and are supported by existing knowledge. This approach allows for the launch of the product while ensuring patient safety, with the understanding that further data and refinement will be undertaken as more information becomes available.
The adoption of patient-centric specifications, which incorporate scientific knowledge and insights gained throughout the development and life cycle of a pharmaceutical product as a standard in pharmaceutical manufacturing of C>s, offers a dual benefit for both industry and regulators. By aligning the objectives of ensuring quality patient care and optimizing manufacturing efficiency and control, patient-centric specifications contribute to a win-win situation for all stakeholders involved.
The concept of reusing well-designed PACMPs, as discussed in ICH Q12, holds particular significance for C>s. These PACMPs can serve as a framework in the absence of specific guidance, providing a general understanding of how comparability will be addressed. Although certain cases may require more specific protocols, having an agreement on broad terms for comparability can still prove to be a valuable tool. This approach promotes efficiency and consistency in assessing and ensuring the comparability of C>s throughout their life cycle. The implementation of a PACMP offers several advantages for applicants. It allows them to:
- Define the level of risk associated with a change or multiple changes in relation to patient safety
- Use their knowledge and expertise to demonstrate how the impact of a change can be assessed, providing insights into the management of residual risks
- Determine appropriate tests to be conducted to evaluate the change, as well as establish suitable boundaries for these tests and provide rationale for their selection
- Reach an agreement with the competent authority on the strategy for managing the change, along with the reporting category for subsequent confirming data. This approach fosters certainty for both the applicant and the reviewer, enabling improved planning and execution of changes throughout the product life cycle
- Facilitate continuous improvement to ensure ongoing patient supply
Conclusion
C>s are a vital and growing part of the modern approach to therapy. The tools outlined in ICH Q12 provide insights into how this post-approval data could be used to facilitate change for these products. In particular, the PACMP could be used to leverage ongoing monitoring data in transparent discussions with agencies and thereby progress changes in an efficient manner, ultimately providing a mechanism to maintain products in a state of control and ensure a continuous supply to patients.
C>s are generally produced in small batch quantities, necessitating the production of multiple batches to adequately support early phase clinical trials. As the product advances to the commercialization stage, the demand for C> batches significantly rises due to data generated from a larger patient pool, thereby providing opportunities to enhance knowledge on the relationship between product quality attributes and clinical outcomes.
The generation of a substantial database during the life cycle management of C>s serves as a crucial resource, enabling the justification of post-approval changes and facilitating the continuous improvement of the product. Moreover, the combination of data from chemistry, manufacturing, and controls (CMC), and patient experience provides compelling evidence that any manufacturing changes do not have an adverse impact on the product.
The establishment of data ecosystems becomes imperative as an extensive volume of data is continuously being gathered, aiming to a future state where correlations between CMC and clinical data can be identified. Implementing suitable data and analytics ecosystems, such as those facilitated by artificial intelligence, will make it feasible to monitor and compare safety, efficacy, and patient benefits with corresponding product quality information. This integrated approach ensures a comprehensive understanding of product performance and supports a high-level analysis to facilitate life cycle management.
The PACMP presents a unique opportunity for collaborative efforts between the industry and regulatory agencies to streamline life cycle management. This initiative aims to identify pertinent CMC and clinical data, fostering the establishment of an effective platform for improved communication between regulators and the industry.
A crucial aspect of this endeavor is to encourage the adoption of industry best practices. This ensures the collection of comprehensive information throughout the product life cycle and establishes strong connections between the data and clinical outcomes. By joining forces, stakeholders can proactively enhance the understanding of product performance, safety, and efficacy, which leads to more efficient and informed decision-making processes.


