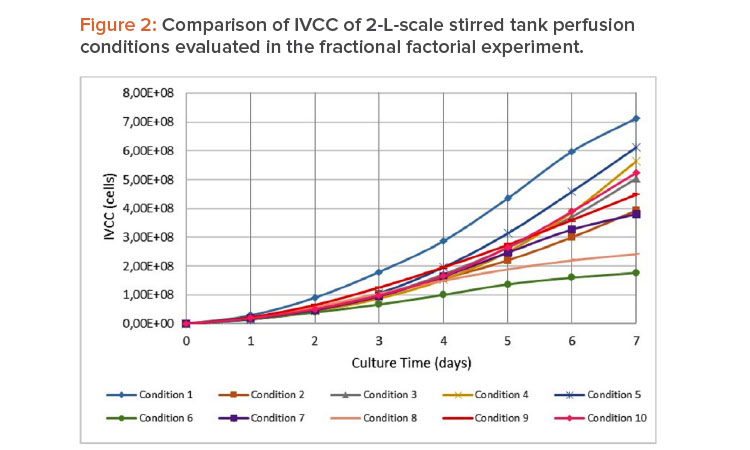
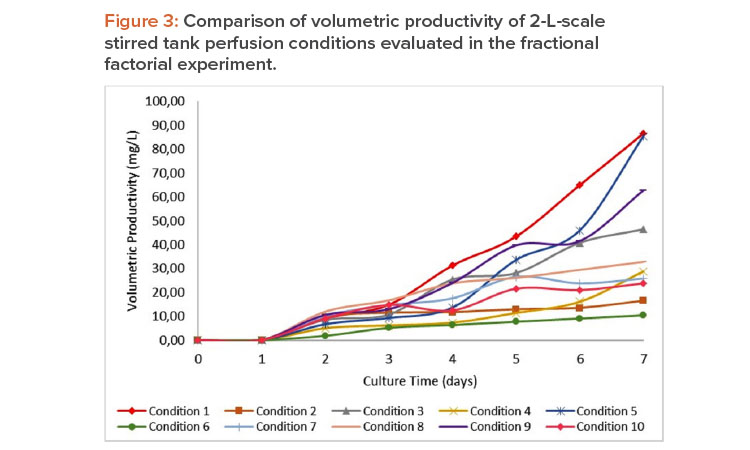
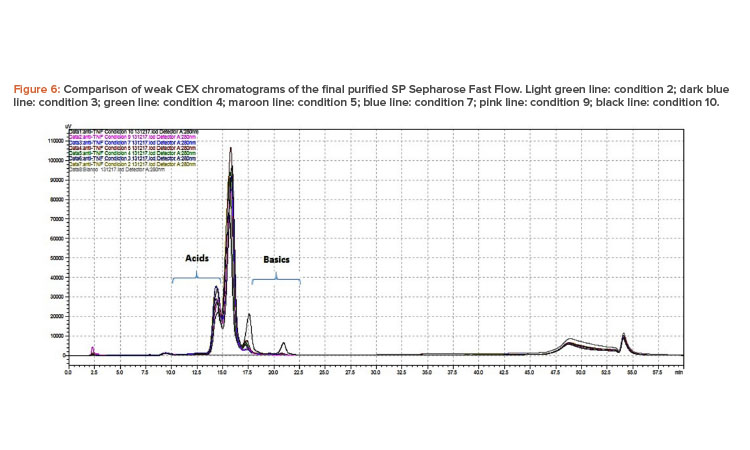
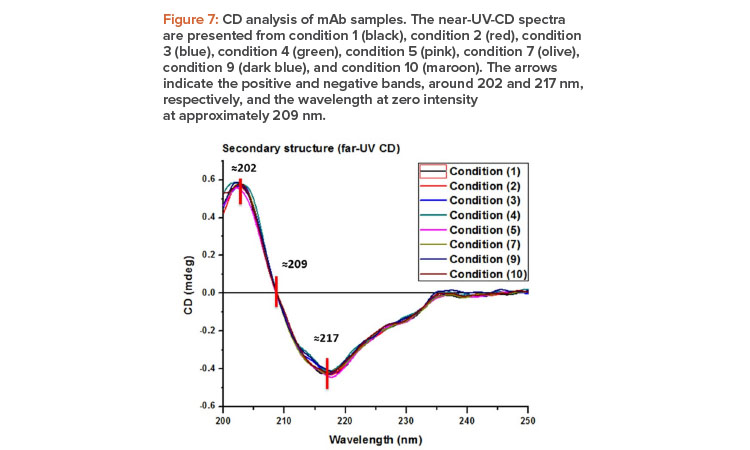
This article describes experiments carried out to evaluate the impact of physical variables of agitated tank fermentation on the process performance and product quality profile of therapeutic mAbs. The aim of these experiments was to develop a consistent fermentation manufacturing process for a therapeutic biosimilar mAb that would maintain the desired quality attributes. Fractional experimental design allows the evaluation of culture media, temperature, airflow, and agitation speed variations on product purity, isoforms composition, secondary and tertiary structure, and ligand binding.
Materials and Methods
Fermentation Process
An NS0 cell line clone producing a chimeric IgG1 mAb was grown in 2-liter (L) stirred bioreactors (Applikon, The Netherlands) in perfusion. The bioreactors were seeded at 5×105 cells/mL, and dilution velocity was increased considering a cell-specific perfusion rate of 0.1 nL/cell/day.
Experiments to elucidate the impact of media and fermentation process parameters were carried out as a reduced two-level factorial with two center points. A commercial media formulation (CMF; a commercially available protein-free hybridoma medium [PFHM-II]) and a mix of CMF and a proprietary media formulation (MB02) 50/50 volume per volume (v/v) were assessed. Impellent tip speed ranged from 0.6 to 1.0 m/s, airflow from 0.0075 air volume per medium volume per min (vvm) to 0.0225 vvm, and temperature from 34°C to 37°C (Table 1); pH was set between 6.8 and 7.2, and the dissolved oxygen concentration upper limit was set to 50% ± 5%.
To monitor cell density, viability, and IgG concentration, 30-mL samples were collected daily. Cells were counted in a Neubauer improved hemocytometer; cell concentration and viability were assessed by the trypan blue exclusion method; and IgG concentration was determined by ELISA.. Perfusion supernatant was filtered through a 3-mm to 0.2-mm filter tandem prior to the purification process.
To set the key process parameter of fermentation operation, a numerical optimization function was made using Design-Expert 6.0.1 software.
Purification Process
Figure 1 depicts the mAb purification process. The mAb preparation was loaded onto a protein A support equilibrated with 200 mM Tris, 100 mM NaCl, pH 7.4 buffer, and eluted from the protein A support with 100 mM glycine, pH 3.0 buffer. The eluate comprising the mAb was buffer-exchanged on G-25 medium. It was subsequently loaded onto a cation-exchange (CEX) chromatography support equilibrated with 50 mM sodium acetate, pH 5.0 buffer; eluted with 28 mM sodium citrate, 200 mM NaCl, pH 7.6 buffer; and finally filtered through a Sartobind Q filter. The absorbance units were measured at 280 nm.
Table 1: Factorial design runs for four factors applied to the development of the process design space of an mAb.
| Condition |
Media |
Stirring,
rpm (m/s) |
Airflow,
vvm |
Temperature,
°C |
| 1 |
CMF |
150.00 (0.4) |
0.0075 |
37.00 |
| 2 |
CMF |
450.00 (1.2) |
0.0225 |
34.00 |
| 3 |
CMF |
150.00 (0.4) |
0.0225 |
37.00 |
| 4 |
CMF |
450.00 (1.2) |
0.0075 |
34.00 |
| 10 |
CMF |
300.00 (0.8) |
0.0150 |
35.50 |
| 5 |
CMF/MB02 |
150.00 (0.4) |
0.0075 |
34.00 |
| 6 |
CMF/MB02 |
450.00 (1.2) |
0.0225 |
37.00 |
| 7 |
CMF/MB02 |
450.00 (1.2) |
0.0075 |
37.00 |
| 8 |
CMF/MB02 |
150.00 (0.4) |
0.0225 |
34.00 |
| 9 |
CMF/MB02 |
300.00 (0.8) |
0.0150 |
35.50 |
Physicochemical Characterization
SDS-PAGE Under Nonreducing Conditions and Western Blot
Sodium dodecyl sulfate polycrylamide gel electrophoresis (SDS-PAGE) is a common technology for antibody-purity analysis. In this case, purified mAbs were resolved on 7.5% polyacrylamide gels under nonreducing conditions followed by silver nitrate staining.
For western blot analysis, proteins were transferred to a 0.45-mm nitrocellulose membrane (Whatman), blocked with 5% skim milk (Fluka) in Tris-buffered saline and probed with goat anti-human-gamma-chain-specific phosphatase alkaline–conjugated antibodies (Sigma) diluted in phosphate-buffered saline (PBS) with 0.1% TWEEN 20. The western blot was developed with Fast Red detection reagent (Sigma).
Size Exclusion Chromatography (SEC)
Chromatographic analyses were performed on a high-performance liquid chromatography (HPLC) system (Shimadzu, Kyoto, Japan). SEC was performed using a TSKgel G3000SWXL column (Tosoh Bioscience, Stuttgart, Germany). Samples (500 μg) were injected, and UV detection was carried out at 280 nm.
Cation-Exchange (CEX) Chromatography
CEX separation was performed using a weak CEX resin (ProPac WCX-10, 4×250 mm, Dionex, Germering, Germany). The mobile phases used were mobile phase A (0.01 M sodium phosphate buffer, pH 6.6) and mobile phase B (0.01 M sodium phosphate buffer + 0.5 M NaCl, pH 6.6). The elution was performed by an ascending gradient from 4% to 80% eluent B before the eluent composition was returned to the starting condition (100% eluent A). UV detection was carried out at 280 nm.