Updated GAMP® GPG Incorporates AI and Open-Source Software

The landscape of clinical trials has been transformed in a post-pandemic world. In July, ISPE released the second edition of ISPE GAMP® Good Practice Guide: Validation and Compliance of Computerized GCP Systems and Data – Good eClinical Practice. It addresses managing complexities associated with decentralized clinical trials, the benefits and challenges of using open-source software, and more.
The conduct of clinical trials and their support by technology solutions has dramatically changed since the first publication of the Guide. The COVID-19 pandemic forced sponsors to rapidly develop solutions so that clinical trials could be conducted during lockdown, which led to the significant adoption and advancement of decentralized trials. This paradigm shift required technical solutions that were patient-friendly, easy to use, fit for purpose, and compliant.
The pandemic also pushed the analysis of real-world data (e.g., from electronic health records) in attempts to find treatment approaches from data collected outside of clinical trials. Both approaches require handling huge amounts of data, necessitating using tools typically associated with processing big data.
In parallel, significant advancements in artificial intelligence (AI) and machine learning (ML) have enabled new approaches for clinical trials, from protocol design to clinical trial closure. This updated guide reflects the content and concepts published in the ISPE GAMP® 5: A Risk-Based Approach to Compliant GxP Computerized Systems (Second Edition) 2. It was time for ISPE to update this key guidance document to reflect technological progress and regulatory advances.
The overall approach, framework, and key concepts as presented in the first edition remain unchanged. However, the second edition advances these concepts to continue promotion of patient safety and data integrity in clinical trials in the post-pandemic world. This is done by facilitating and encouraging the achievement of computerized systems that are effective, reliable, and of high quality. This article provides an overview of the Guide’s update and discusses a selection of four major topics related to the innovations in the second edition.
Background and Drivers
In recent years, a transition from the traditional, site-centric model to a hybrid or participant-centric decentralized model has become more prevalent. This transition was supercharged by the COVID-19 pandemic. However, the transition to more decentralized models started even earlier. The aim was to reduce cost, enhance data accuracy, improve participant access and engagement, overcome geographic barriers, and promote diversity.
Today, virtual or decentralized models have gained traction as clinical trial computerized system applications enable remote data collection and virtual monitoring. Regulators have reacted to this and issued guidance for decentralized models, such as the US Food and Drug Administration’s (FDA) guidance on “Conducting Clinical Trials with Decentralized Elements” 3.
Decentralized clinical trials (DCTs) led to an increase in the development and usage of wearables and sensors within clinical trials. These technologies produce vast amounts of data, as they are continuously monitoring parameters and recording data that are important for a clinical trial. The management, evaluation, and analysis of this data requires approaches and tools by which clinical trial teams can extract insights from large amounts of data. As such, manual review, control, and assessment by humans is becoming infeasible. Still, these approaches need to be fit for the purpose. Therefore, data management in clinical trials has evolved into data science and is now adopting advanced approaches including the use of AI methods, ML in particular.
Figure 1: Overview of clinical project layers
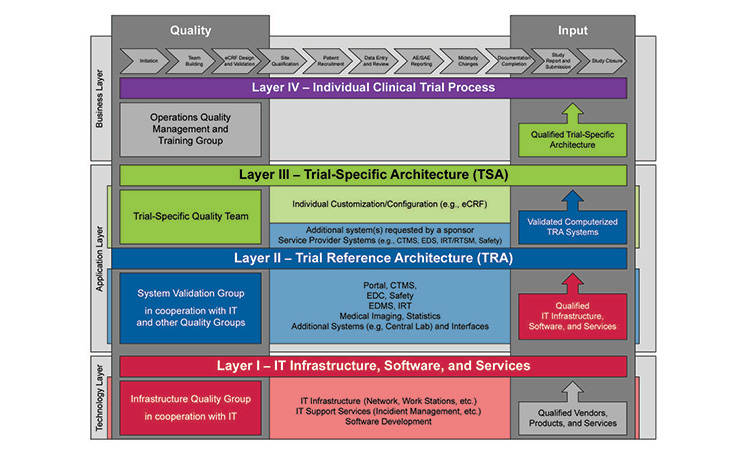
Source: International Society for Pharmaceutical Engineering. ISPE GAMP® Good Practice Guide: Validation and Compliance of Computerized GCP Systems and Data – Good eClinical Practice (Second Edition). North Bethesda, MD: International Society for Pharmaceutical Engineering, 2024.
Similar amounts of data from various sources in the real-world context are evaluated by researchers to gain new insights or leads and to support controlled clinical trials. The quality of such real-world data, their management, and further analysis is of key importance to allow the generation of real-world evidence. The impact of biased data and inappropriate methodology proposes a significant risk to the safety of patients. This is highlighted by the COVID-19 hydroxychloroquine study published in The Lancet that had to be retracted because the findings were based on electronic health record (EHR) data from inconsistent sources, compromising the overall quality of the combined data set 4, 5.
All real-world data and data collected, used, and analyzed in clinical trials originate from humans. This data is typically of a highly sensitive nature because it is concerned with medical conditions, treatments, and lifestyles. Therefore, the protection of data privacy is paramount when conducting clinical trials using real-world evidence and in the management of associated real-world data.
The outsourcing of clinical trials and supporting technology has progressed further since the first edition’s release. Almost all sponsors and clinical service providers routinely use the cloud-based Software-as-a-Service (SaaS). The regulators have issued guidance documents, including the European Medicines Agency’s (EMA) “Guideline on Computerised Systems and Electronic Data in Clinical Trials” 6.
This guidance clarified the quality expectations and roles and responsibilities in the usage of cloud service providers, such as SaaS. Additionally, it also clarified the quality expectations on clinical site operated systems that are used to generate, collect, or analyze data during a clinical trial. Furthermore, the guide needed to be aligned with the concepts and principles of ISPE GAMP® 5 (Second Edition) 2. This included emphasis on critical thinking, testing of computerized systems, and guidance on outsourced IT infrastructure or systems.
GAMP guidance must stay fully aligned with current good practice to provide optimal value to the industry, avoiding guidance based on outdated technical concepts, approaches, or techniques, even if such concepts, in some cases, remain in regulatory guidance or company policies and procedures. In light of current developments as described in this article, an update of the ISPE GAMP® Good Practice Guide: Validation and Compliance of Computerized GCP Systems and Data – Good eClinical Practice 1 was necessary.
Overview: Framework and New Concepts
The updated Guide contains the same approach and key concepts, including the Clinical Project Layer Model, from the first edition (see Figure 1). It also provides the following:
- Updated overview of current regulatory guidance documents
- A high-level process overview, including the Clinical Project Layer Model
- Relevant data flows
- Required oversight activities
- Inspection readiness
- A risk-based approach for the setup of clinical systems
Each process step—from planning, conduct, and analysis to reporting—is analyzed for its potential risks to patient safety and the data integrity of the potential supporting technologies. Then an outline is formed for a potential validation approach for these technologies. The life cycle of clinical data and the associated data integrity risks are analyzed, and approaches for mitigating these risks are presented. This includes specific guidance for electronic source data, audit trails and audit trail reviews, interfaces and their validation, electronic and digital signatures, and certified copies.
The newly created appendices provide practical guidance on (1) data privacy in clinical trials, (2) the specific aspects to be considered in decentralized clinical trials (DCTs), (3) good clinical laboratory practice, (4) the use of data science and AI-enabled systems, (5) real-world data, (6) real-world evidence, and (7) the open-source software used in clinical trials. The Guide emphasizes that critical thinking should be applied through the trial, data, and system life cycle and is fully aligned with the principles and concepts of ISPE GAMP® 5 (Second Edition) 2.
Data Science and AI in Clinical Trials
Data science disciplines, including the use of AI and ML, offer new ways to analyze data, extract information and patterns, form predictions, create recommendations, and generate content such as text and images. As such, clinical processes may be augmented with AI. AI approaches can improve efficiency of clinical trials and support success and quality of clinical trial outcomes. Especially where a high volume of data is generated, and several possible paths and choices are evaluated, classical ways of quality assurance, analysis, and assessment hit limits of efficiency and practicability. However, the statistical nature and the complexity of these approaches comes with their own risks, as is the even higher dependency on high-quality data and an understanding of representativeness of data for a given application, both for data science as well as AI approaches 7, 8.
ISPE GAMP® 5 (Second Edition) appendix D11 offers a life cycle model focused on ML subsystems and the three life cycle phases of concept, project, and operation with good ML practices and operations (MLOps) 2. These include concepts such as splitting data for training, validation, and testing purposes as well as an iterative fine-tuning approach for the final tailoring of the model, toward acceptance, release, and ongoing monitoring. In addition, the ISPE GAMP® 5 (Second Edition) appendix D11 elaborates on supporting processes, including risk management, data governance, and change management. This underpins the relevance and importance of data and the forward-looking perspective of careful evaluation of risks to patient safety, product quality, and data integrity.
Although ISPE GAMP® 5 (Second Edition) appendix D11 provides an overarching framework, this guidance needs to be interpreted and tailored for specific use cases in the clinical trial context 2. To serve this goal, the data science and AI chapter in the ISPE GAMP® Good Practice Guide: Validation and Compliance of Computerized GCP Systems and Data – Good eClinical Practice (Second Edition) elaborates on specific aspects that are relevant in clinical trials.
Suitability Assessments
The innovative and project nature of clinical trials requires a thorough assessment of the suitability and fitness for purpose in use of data science and AI approaches. Such assessments are performed during the concept phase of the computer system development and part of a dedicated trial.
Diligence
Clinical trial protocols require diligence, not only in terms of patient safety, but also commercial viability regarding the use of AI, ML, and data science approaches.
Patient Data
Clinical trial activities require close interaction with patient data in various process steps (e.g., recruiting, data capture, and evaluation) and potential areas of direct interaction with patients via digital health technologies. Therefore, a thorough understanding of the stakeholder ecosystem is crucial for successful use of AI in the clinical context.
Guidance Provided
Considering these aspects, the chapter on data science and AI in the new guidance document provides detailed guidance tailored to supporting clinical trial operations. In particular, the chapter provides dedicated guidance regarding the following themes:
- General definitions and an overview on regulatory standards with relevance to data science, AI, and ML concepts
- The evolution of clinical data management into clinical data science and data analytics approaches with the application of emerging technologies
- Guidance on AI-/ML-enabled systems across use cases along the clinical trials process, to provide overarching guidance on the life cycle management decisions, considerations on fitness for purpose, and importance of human–machine interaction concerning a variety of stakeholders in the clinical trial ecosystem. Example use cases illustrate potential risks to consider, risk control, and validation strategies throughout the clinical trials process
This chapter should support the safe and effective adoption of data science, AI, and ML approaches in the clinical context. These are integrated with further cross-sectoral operational guidance. In particular, this guide was developed with the focus of Good Clinical Practice (GCP) based on concepts included in ISPE GAMP® 5 (Second Edition) 2.
Real-World Data and Evidence
Real-world data (RWD) and real-world evidence (RWE) have been integral in epidemiology for years, but their significance in healthcare and pharmaceutical research has surged recently. Regulatory agencies like the US FDA and the EMA now recognize the value of RWD and RWE for evaluating medical product safety and efficacy. These bodies have developed frameworks to facilitate the incorporation of RWE in regulatory decision-making processes. Mistakes in the selection and processing of RWD have significant impact on the safety and well-being of patients, as seen in the retracted article in The Lancet article 4, 5. For this reason, the Good Practice Guide covers the following themes:
Key uses of RWD and RWE include:
Clinical trials:
- Historical control group or concurrent control group
- Patient recruitment, site selection and feasibility assessment analyzing demographic data, treatment patterns, and healthcare utilization from RWD sources, researchers can identify sites with a sufficient patient pool for specific clinical trial criteria
- Patient population analysis to provide insights into the characteristics and distribution of patient populations in different geographical regions
Post-marketing:
- Post-marketing surveillance to enable continuous monitoring of drugs and medical devices/digital health technologies after they enter the market
- Post-market effectiveness studies to assess the effectiveness of drugs after they have been launched in the market
- Label expansion and regulatory submissions support label expansion efforts by providing additional evidence on drug effectiveness, safety, and efficacy in broader patient populations or new indications
- Evidence generation for the safety, efficacy, and effectiveness of interventions in diverse patient populations and real-world settings
- Comparative effectiveness and safety studies to compare the effectiveness and safety of different drugs or treatment interventions in real-world clinical settings
- Health technology assessment to support evaluations of pharmaceutical products providing evidence on the comparative effectiveness, safety, and cost-effectiveness
- Treatment access and reimbursement use RWE to assess the value of therapies, make coverage decisions, and determine appropriate pricing
Personalized medicine and biomarkers:
- Personalized medicine and precision health (e.g., identification of patient subgroups, biomarker analyses, and treatment response assessments)
- Identification of biomarkers that are associated with specific clinical conditions, outcomes, or treatment responses
Challenges Using RWD and RWE
Data quality and standardization
RWD is derived from various sources, such as EHRs, claims databases, patient registries, and even wearable devices. These sources often have inherent limitations, including missing data, inconsistencies, and variations in data collection practices. Ensuring data quality and achieving standardization across different sources is a significant challenge. High-quality, standardized data are crucial for generating reliable and actionable RWE.
Data privacy and security
RWD frequently contains sensitive patient information, raising concerns about privacy and security. Robust anonymization and stringent security measures are essential to protect patient privacy while leveraging RWD for research purposes. Balancing the need for data access with privacy protection is critical to maintaining public trust and complying with legal and ethical standards.
Bias and confounding
RWD can introduce biases and confounding factors that may affect the validity and reliability of research findings. Unlike randomized controlled trials, real-world studies cannot always control all variables, making it challenging to isolate the effects of the intervention being studied. Researchers must develop and apply sophisticated statistical methods to account for these biases and confounders to ensure credible RWE.
Reproducibility and transparency
For RWE to be trusted, the methods used to generate it must be transparent and reproducible. Research findings should be independently validated and reproduced to build confidence in RWD and RWE studies. This requires clear documentation of methodologies, data sources, and analytical techniques, allowing other researchers to replicate the studies and confirm the results.
Regulatory and reimbursement acceptance
Although regulatory bodies like the US FDA and EMA have recognized the value of RWD and RWE, consistent industry standards and methodologies for generating RWE are still needed. These standards are essential to ensure that RWE is acceptable for regulatory and reimbursement decision-making. Establishing clear guidelines and frameworks that provide consistency and clarity in the generation and use of RWE remains an ongoing challenge. Regulatory and reimbursement bodies must work together with industry stakeholders to develop these standards and facilitate the integration of RWE into decision-making processes.
To address these challenges the Good Practice Guide 1 provides a process for evaluating and using RWD to generate RWE, which covers:
- Determination of the intended use based on a defined research question
- Data accrual to determine the required data and their quality criteria
- Selection and assessment of data sources that can provide the required data
- Data preparation and curation, including data cleaning, usage of a common data model, and data standardization
- Management and assurance of data privacy aspects
- Data management throughout the entire data life cycle
- Data retention requirements based on usage and applicable regulations
With clinical trial computerized system applications enabling remote data collection and virtual monitoring, decentralized models have become more efficient from a technology point of view.
The detailed guidance given for each of these steps in the Guide ensures data integrity and data quality that is sufficient for the intended use of the RWE generated from RWD.
Decentralized Clinical Trials
Multiple clinical trial delivery models are used in the pharmaceutical industry. In recent years, a transition from the traditional, site-centric model to a hybrid or participant-centric decentralized model has become more prevalent. Drivers from centralized to more decentralized models include cost and limited engagement of some trial participants due to geographic and socioeconomic barriers. With clinical trial computerized system applications enabling remote data collection and virtual monitoring, decentralized models have become more efficient from a technology point of view.
According to the US FDA guidance document “Conducting Clinical Trials with Decentralized Elements,” published in September 2024, DCT is defined as a clinical trial where some or all of the trial-related activities occur at locations other than traditional clinical trial sites 3. A DCT model brings all or part of the clinical trial to the patient, such as to their home, a local healthcare facility, or a laboratory, thus reducing some of the challenges for trial participants. With a DCT, the goal is to minimize the patient burden while collecting real-time, accurate clinical trial data. While the concepts and principles of validation are consistent across GxP areas, there are key areas where additional considerations are needed to support these technologies 3.
When transitioning from traditional to a decentralized or hybrid clinical trial delivery model, the readiness of the organization’s infrastructure and workforce must be considered. Integrating systems to support remote monitoring, data collection and training for the organization, clinical site staff, and patients may be needed. As with all aspects of clinical trials, regional and local regulatory compliance requirements should also be considered.
Decentralization introduces further complexities to various processes in clinical trials, including monitoring, audits, privacy and blinding, and business continuity. For instance, where the participants receive investigational medicinal products (IMPs) directly, additional focus should be placed on the expiration date. Furthermore, patient safety is a concern where access to bulk IMP may be available for the trial participant.
Reflecting on these challenges, this new chapter provides guidance regarding:
- Key prerequisites to be considered when implementing DCTs, including the business process flow, data flow, technical needs, and site feasibility
- Supplier relationship considerations and the need to ensure that contractual agreements and service level agreements are in place
- Ensuring mechanisms for data control throughout the clinical trial to maintain data integrity
- Use of digital health technologies and devices including provisioning, support, access, and safety and biohazard concerns
- Establishing fitness for intended use of a potentially complex clinical trial setup, including activities for infrastructure qualification, software development, and operational support and maintenance
- The importance of developing a dedicated training program for participants, investigators, clinical trial staff, sponsors and other parties, including training materials and the delivery method
- Considerations regarding shipment of IMPs or investigational products direct to patient or direct to site, and storage and disposal needs
Good Clinical Laboratory Practice
In clinical trials, analyzing samples from participants is crucial for proving drug effectiveness and ensuring participant safety. These samples are examined based on parameters outlined in the clinical trial protocol. Effective tracking and handling are vital to maintain data accuracy and completeness. Samples are often analyzed away from the collection site, necessitating strict control over their chain of custody, including time, environmental conditions, and transportation routes. Adequate training for staff at the collection site ensures proper sample labeling and handling, with blinding maintained throughout all stages.
Mapping the Business Process and Data Flow
To establish the necessary technical, procedural, and behavioral controls for sample and data integrity, it’s essential to map out the business process and its dependencies. This involves understanding the computerized systems and responsible parties involved, from sample collection to result reporting. Particular attention should be given to system interfaces and the data life cycle (i.e., transfers, archiving) to ensure these systems are well-specified, tested, controlled, and fit for purpose. The integrity of the data must be maintained throughout the life cycle.
Risk Evaluation and Documentation
Understanding the business process and data flow allows for evaluating and documenting risks associated with the process. Key risk considerations include:
- Unavailability of source or target systems within a data flow
- Corrupted or incomplete laboratory data related to participants
- Loss of association to a participant number
- Unblinding issues depending on the trial design
- Inadequate control of third parties
Decisions should be based on risk assessments using critical thinking. Ensuring computerized systems and data transfers are specified, tested, and controlled is essential for data integrity.
Roles and Responsibilities
Samples may be analyzed by the sponsor company or third parties, including investigator sites, clinical service providers, central laboratories, hospital laboratories, or university laboratories. All partners performing sample analysis must be qualified, with responsibilities clearly defined in contracts. This clarity ensures accountability and proper management of the sample analysis process.
Regulatory Guidance
The EMA’s “Guideline on Computerised Systems and Electronic Data in Clinical Trials,” published in March 2023, outlines the necessary controls to ensure data integrity throughout the data life cycle 6. This guidance addresses technical, procedural, and behavioral controls to maintain data accuracy and completeness in clinical trials.
Good Clinical Laboratory Practice
In clinical trials, analyzing samples from participants is crucial for proving drug effectiveness and ensuring participant safety. These samples are examined based on parameters outlined in the clinical trial protocol. Effective tracking and handling are vital to maintain data accuracy and completeness. Samples are often analyzed away from the collection site, necessitating strict control over their chain of custody, including time, environmental conditions, and transportation routes. Adequate training for staff at the collection site ensures proper sample labeling and handling, with blinding maintained throughout all stages.
Quality Management Systems and Laboratory Standards
Laboratories analyzing clinical trial samples must be qualified, with all interfaces to laboratory systems validated. The EMA’s “Reflection Paper for Laboratories that Perform the Analysis or Evaluation of Clinical Trial Samples,” published in February 2012, details the quality system elements necessary for such laboratories, including procedures for emergency release of safety-impacting results 10.
Standard Controls and Documentation
Controls necessary to support sample analysis in clinical trials are standard requirements within quality management systems. These include policies and procedures for environmental controls, sample labeling, storage, transport, data transfer, review and release of sample analysis, maintaining the blind, sufficient resources, appropriate training, contracts, agreements, and quality assurance. Both the ISPE GAMP® Good Practice Guide: GxP Compliant Laboratory Computerized Systems (Second Edition) and ISPE GAMP® 5 (Second Edition) provide information on ensuring computerized systems are fit for purpose. They also safeguard patient safety, product quality, and data integrity 2, 11 .
The updated Good Practice Guide provides practical guidance criteria for selecting partners involved in the analysis of the samples. The requirements for the logistics and the analysis of samples are explored and a typical workflow for laboratory is outlined. Typical challenges in the implementation and validation of supporting computerized systems for this process are outlined. This includes systems such as laboratory information management systems and also addresses the data transfer between partners and the necessary retention and archiving of laboratory records. The need for clearly defined responsibilities, adequate facilities, and processes is explored in detail.
Conclusion
The principles of ISPE GAMP® 5 (Second Edition) needed to be interpreted for computerized systems used in clinical trials to provide practical guidance to validation experts that are challenged with the validation of these systems. The high degree of outsourcing, the cross-organizational usage of many of the systems, and the various regulatory aspects (including data privacy) provide unique challenges in the validation of GCP systems.
This article only outlines a few sections of the ISPE GAMP® Good Practice Guide: Validation and Compliance of Computerized GCP Systems and Data – Good eClinical Practice (Second Edition). As of today, it is the most comprehensive guide on the validation of computerized systems used in the context of drug development and clinical trials available.
About the Authors







