Live Biotherapeutic Products: Moving the Microbiome to the Patient

Live biotherapeutic products (LBPs) have the potential to treat a wide range of ailments. However, these living microorganisms are difficult to produce due to evolving government regulations and limited GMP manufacturing experience. New facility designs and more specific process guidance could help overcome these challenges. This article explores the nuances of facility design and regulatory requirements for the development of LBPs.
The human microbiome is composed of a diverse community of microorganisms that varies by person based on both genetic and environmental factors. As evidence emerges linking the human microbiome to health and disease, interest in LBPs has followed. These products have the potential to treat a wide range of ailments from cancer to autoimmune conditions.
The US FDA regulatory framework defines LBPs as biological products that contain live organisms (bacteria or yeast) that are intended for the treatment or prevention of disease.1 One or multiple microbial strains or genetically engineered microorganisms can be used for the production of live cells in LBPs. As defined by the FDA, LBPs exclude vaccines, filterable viruses, oncolytic viruses, and products intended as gene therapy agents.
LBPs are living products and complex to produce. The typical manufacturing process involves fermentation starting from carefully selected cell banks, separation, formulation, and filling followed by lyophilization. Facility design can involve spore-forming organisms, which can require additional containment and biosafety considerations over standard biologics facilities.
Microbiome Market
In November 2022, the FDA approved the first microbiota-based LBP, and the Australian health authority, Therapeutic Goods Administration (TGA), allowed the debut of microbiome-based therapy. These products are approved for the treatment of recurrent C. difficile infections, a condition that causes on the order of 30,000 deaths each year in the United States alone.2 The Centers for Disease Control and Prevention (CDC) classified C. difficile as a top microbial threat to human health in their report, “Antibiotic Resistance Threats in the United States, 2019.”3 Many other products are in research and development and in various clinical phases. Given this trend, approval and commercial manufacturing of LBPs are imminent.
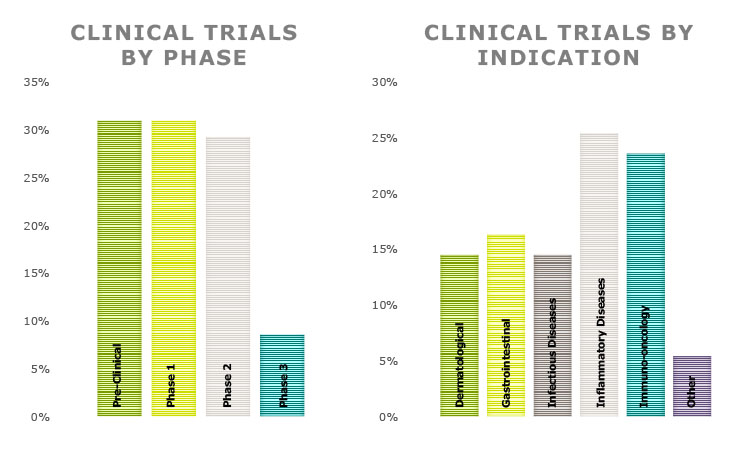
Although access to microbiome products is limited, with few products at this stage, scientific research on the microbiome continues to highlight new findings and will pave the way for more development and advancement. Microbiome treatments are not limited to infectious diseases. The applications being investigated are varied and range from autoimmune conditions and inflammatory diseases to immuno-oncology applications. Figure 1 provides some data on the range of products in various phases of research and clinical trials as well as intended applications for prospective therapeutics.
LBP Manufacturing and Equipment Considerations
Microorganism selection begins with each developer identifying strains that deliver a desired beneficial effect to target a specific disease. LBPs may be composed of a single strain; however, multiple strains, also called a consortium, are necessary for some products for proper therapeutic effect. Although a consortium may provide optimal therapeutic benefit, it entails additional manufacturing challenges because growth of up to 50 unique strains may be required for a single product.
Although the fundamental approach of LBP manufacturing processes is not different from other biopharmaceutical therapies, it is important that the manufacturing process is defined with extensive expertise to achieve critical attributes relating to identity, strength, quality, purity, and potency of selected or engineered bacterial strains for target therapeutic effect.
At a high level, microorganisms for manufacturing LBPs can be classified according to their control requirements for molecular oxygen. Traditional biologics manufacturing has focused on the production of aerobic organisms and cannot accommodate the production of anaerobes. Due to the dominance of obligate anaerobic bacteria in the human gut microbiome, along with better stability and viability, many LBP developers choose either facultative anaerobes or strict anaerobes to develop and manufacture LBPs.
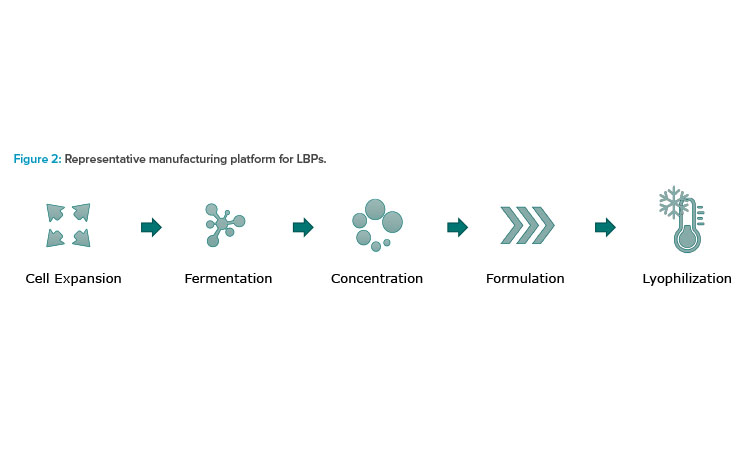
Facultative anaerobes can grow in the presence or absence of oxygen, and obligate anaerobes can only grow in the absence of oxygen, a major consideration for production control. The pillars of microbiome manufacturing platform are similar to a typical biological manufacturing process with fermentation, separation, and lyophilization steps. Figure 2 illustrates a representative manufacturing platform for LBPs.
Defining a scalable manufacturing process is a challenge for LBPs because they are novel medicines with limited GMP manufacturing experience. Compliance with an evolving regulatory framework is another hurdle to overcome.
The production process for LBPs begins with preparation of target frozen cells from the working cell bank. Inoculation takes place within an enclosed system depending on the type of strain and sensitivity to environment. Seed fermentation for cell expansion continues until the target density is achieved and it is transferred to a production fermentor. This is crucial for a scalable biological manufacturing process.
Typical production microbial fermentor sizes range from 500 L to 5,000 L in volume. Both single-use and stainless steel options exist for fermentation. Technology platform selection depends on several factors. Initial setup has differences between single-use and stainless steel, but there is high similarity in the basic method of operation, capabilities, and features.
Stainless steel fermentors are often preferred because they can grow cells to a greater cell density due to their ability to optimize oxygen mass transfer and remove excess metabolic heat. Stainless steel fermentors are also an ideal choice when product dosage and patient population necessitate larger volume production because they offer options that scale up to 5,000 L. Other stainless steel technology benefits include increased resilience to supply chain fluctuations and lower operating costs.
Single-use fermentors are another viable option and can be preferred when the intended patient population or dosage is lower, making smaller-scale production runs a viable option. Single-use fermentors range from 50 L to 300 L, which may require multiple production fermentors operating in parallel if larger volumes are required. The inherent benefits of single-use technology include robust contamination control, operational flexibility, reduced changeover, and reduced process downtime. The elimination of complicated clean-in-place (CIP) and steam-in-place (SIP) cycles and cleaning validation are also favorable.
Downstream concentration is designed in consideration of the fermented microbial broth and the target biomass that unit operations can accommodate to deliver efficient biomass processing either in a batch or continuous setup. The concentration of the cell broth varies depending on the production method and the type of cell strains; therefore, the optimal selection of concentration processing equipment is critical to retain the target cell solid in a reproducible manner.
After fermentation and concentration, the product is formulated with sterile buffers and cryopreservatives. At the conclusion of formulation, the product is lyophilized. Lyophilization is conducted to dehydrate the live biotherapeutic substance and gain stability in the final substance. Filling and lyophilizer loading must take place within a Grade A (ISO 5) designed environment with appropriate containment to avoid contamination of the product or surrounding space. The use of barrier isolator technology may be employed for contamination control and containment and to maintain a reduced oxygen environment if necessary for the product.
After lyophilization, the product is transferred to an intermediate bulk container (IBC). Milling of the product may be required to achieve a specific particle size range and is followed by a blending process. Additional excipient materials are added, and the IBC is rotated to mix the powder. After blending, the IBC is transferred to an encapsulation machine. Although LBPs are researched and developed in various forms, current trends show that capsules are the dominant form within the microbiome space. Oral delivery of LBPs should endure acidic conditions and prevent release of the drug product until it reaches the target intestine. Securing enteric protection is also a critical element for process and product development.
The encapsulation process involves transferring the product into a machine that precisely doses the blended product into gelatin capsules. The machine places a gelatin cap and closes the capsule. Encapsulation systems can include integrated check weighing and metal detection. Capsules are collected in a bin or tote in preparation for final packaging. Product containment during blending and encapsulation should be designed to avoid contamination of the product and the surrounding room.
Manufacturing Facility Considerations
The manufacture of LBPs necessitates distinctive facility design considerations to account for enhanced biosafety levels, manufacture of spore-forming organisms, and multi-strain production.
Spore-Forming Organisms
Spore-forming microorganisms are preferred or chosen for product research and development due to the ability of spores to survive passage through the acidic conditions of the gastrointestinal tract. However, when transitioning to large-scale manufacturing, tough questions must be addressed to control the risk of cross-contamination.
The formation of spores allows for bacteria to survive under adverse environmental conditions, including extreme temperatures, dryness, chemical agents, and even ultraviolet radiation. The Bacillus and Clostridium species are among the LBPs that are spore formers. Dormant spores can survive for many years; when conditions are favorable, the spores can germinate to an active or vegetative state.
Given the robustness of spore-forming organisms, careful consideration must be given to the facility design for the manufacture of these species. The FDA has published “Guidance for the Industry on Manufacturing Biological Intermediates and Biological Drug Substances Using Spore-Forming Microorganisms.”4 Although previous regulations mandated that work with spore-forming organisms be performed in a separate building from non-spore-forming organisms, the latest guidance allows for greater manufacturing flexibility. When manufacturing spore-forming organisms in a multiproduct facility, the paramount concern is prevention of cross-contamination due to the persistent nature of spores. This can be accomplished by a combination of physical containment (equipment and facility design) and procedural controls.
Per the regulatory guidance, double airlocks should be employed for incoming personnel, materials, and exiting waste. Figure 3 illustrates the preferred manufacturing suite arrangement: unidirectional flow of personnel and materials with segregated double airlocks for incoming and exiting personnel and materials. The interior airlocks are negatively pressurized to surrounding spaces to enhance containment. Single-pass (100% outside) air is also recommended for spore-forming product spaces.
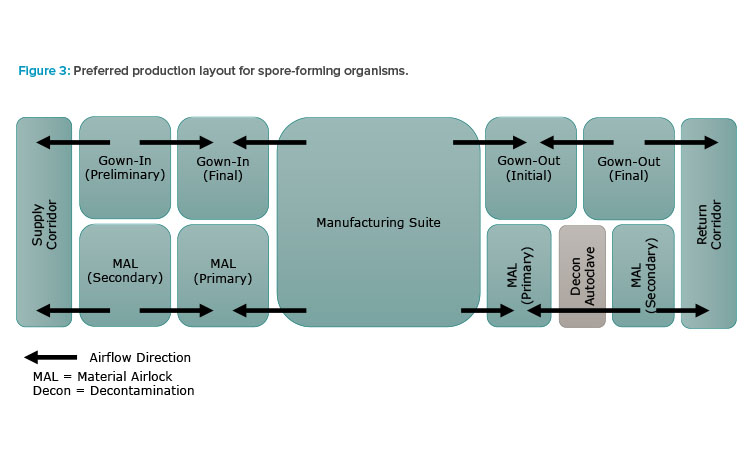
Equipment should be dedicated if possible and the use of single-use equipment is encouraged. If equipment is used on a campaign basis for both spore-forming and non-spore-forming organisms, it must be decontaminated via a validated changeover procedure between campaigns. Manufacturing suites must also undergo a validated decontamination procedure. Use of a gaseous or vapor-phase sterilant, such as vapor-phase hydrogen peroxide (VPHP) or chlorine dioxide gas (ClO2), should be considered and will be discussed in further detail.
Recommended procedural controls include multiple stages of gowning and de-gowning of personnel to minimize the risk of spore-forming organisms exiting the production areas. Personnel who work in spore-forming areas are also encouraged to shower prior to working in other areas of the facility.
Biosafety Level Considerations
Biosafety level design considerations are defined in the US by the NIH in “NIH Guidelines for Research Involving Recombinant or Synthetic Nucleic Acid Molecules” and by the CDC in “Biosafety in Microbiological and Biomedical Laboratories (BMBL), 6th Edition.”5, 6 With increasing biosafety levels, greater protective measures are implemented to protect both the operator and the environment from potentially adventitious agents. Most LBPs are classified as biosafety risk group 2 (BSL-2 or BL2) because they are pathogenic or infectious organisms that pose a moderate health hazard. Additionally, per the NIH, most LBP facilities are classified as “large scale” due to working with volumes of greater than 10 L per container.
LBP facilities are often designated as “BSL-2 enhanced,” or BSL-2+. Although this is not an officially recognized classification by either the NIH or CDC, it indicates that all the requirements for BSL-2 are implemented, along with select BSL-3 level requirements as an added means of protection.
Vapor-Phase or Gaseous Sterilization
Although not a regulatory requirement, many LBP manufacturers incorporate either portable or integrated gaseous or vapor-phase sterilization into their facility design. There are many commercially available sterilants; however, the most commonly used in spore-forming facilities is vapor-phase hydrogen peroxide (VPHP). Gaseous or vapor-phase sterilants are used to destroy all microorganisms that are potentially contaminating a given space. These sterilants are all oxidizers that work by disrupting the cell membrane or cell walls of microorganisms, which in turn leads to cell lysis and death.
Gaseous or vapor-phase sterilants may be used on a periodic basis for campaign changeover and for emergency decontamination in case of a product spill or breach. There are options to integrate this fumigation system fully into the facility design or to contract out the sterilization on a periodic basis using portable units. When considering use of a gaseous or vaporized sterilant, the materials of construction for the equipment and manufacturing space should be evaluated, including whether HVAC ducting will be included as part of the sterilization process.
Not all materials are compatible with fumigation chemicals commonly used in the pharmaceutical industry, including chlorine dioxide, hydrogen peroxide, and formaldehyde. Sterilization chemical selection should be evaluated with regard to multiple different factors, including the spore-forming organisms that are expected to be manufactured or present in the facility because some sterilants are more effective at destroying certain species than others.
Regulatory Considerations
As there is continuous effort to improve the clarity on regulatory requirements for microbiome-based products and LBPs, it is imperative that product development should be conducted in good communication with regulatory authorities. In the US, LBPs are regulated by the FDA through the Center for Biologics Evaluation and Research (CBER). In 2016, the FDA issued a guidance document for early phase clinical trials of LBPs.1
In the EU, LBPs are considered biological medicinal products and are regulated under guidelines for other biological products such as vaccines. In addition, guidance specific to LBPs has been developed. In 2019, the EU released a monograph addressing LBPs for human use.7
It is important to note that the guidances issued to date are primarily focused on requirements for clinical trials, with little advice available for commercial drug regulatory requirements due to the developmental nature of LBP manufacturing. For an Investigational New Drug (IND) application, both the FDA and EMA expect specific information and guidance requirements are in place for both drug substance and drug product. The CMC requirements for drug substance products for LBP INDs are (a) the LBP description, including strain designation, source, phenotype, and genotype; (b) the LBP’s characterization, including antibiotic resistance profiles; and (c) the method of manufacture, including raw materials, production flow chart, cell banking, cell growth and harvest, purification, and in-process testing. The CMC requirements for drug products for LBP INDs are the composition; manufacturer; specification, including identity, potency, purity, and microbial bioburden; stability; placebo; and environmental assessment.
Conclusion
Although there are few commercially approved LBPs to date, the clinical pipeline shows a number of products with promise and a commercially approved product is likely in the near future. The manufacture of LBPs is unique from both an equipment and facility design perspective due to the requirements of facultative and strict anaerobes, spore-forming organisms, and because the end product is a living organism. Biosafety considerations and the implementation of facility controls to maintain containment requirements are crucial. Both the FDA and EMA have issued regulatory guidance, but it primarily focuses on CMC requirements for IND applications, with less focus on how drug regulatory requirements are to be addressed in practice.


