Design Considerations for Large-Scale Stem Cell Manufacturing

There is much that large-scale commercial stem cell therapy processes can adopt from the existing bioprocessing industry. This article addresses some of the unique challenges posed by large-scale stem cell and stem cell–derived product manufacturing processes, and what should be considered while designing a manufacturing facility.
In 1931, Swiss surgeon Paul Niehans injected parathyroid cells from a calf embryo into a woman whose parathyroid gland had been partially removed by accident during surgery. Though his claims of multiple successful treatments of cancer using this technique have never been validated by research,1 his idea of using live cell products to treat patients spawned an industry that has produced many scientifically proven products.
As of Q2 2022, there were 59 nongenetically modified cell therapies approved for commercial use and an additional 803 in development globally.2This excludes ex vivo gene therapies such as chimeric antigen receptor T cells (CAR Ts) or T cell receptors (TCRs), which are often lumped together as “cell therapies.” Nongenetically modified cell therapies generally fall under two umbrellas: autologous and allogeneic. Autologous therapies use a patient’s cells as the starting material, whereas allogeneic therapies use donor cells.
There is a strong desire in the industry to move toward allogeneic cell therapies because they have several advantages over autologous cell therapies, including reduced cost to the patient, ease of automation, and ability to produce scalable “off-the-shelf” products. Nevertheless, allogeneic cell therapies face challenges to reaching commercial viability, such as the potential to induce graft-versus-host-disease (GVHD) and the risk of immune-mediated rejection by the host. Large-scale allogeneic stem cell manufacturing will become increasingly common as these challenges with GVHD are addressed, as the industry matures, and as indications with larger patient populations are targeted.
When it comes to manufacturing scale, allogeneic cell therapies currently being developed and manufactured are based on different modalities, which dictate batch size. Allogeneic CAR-Ts and natural killer (NK) cells are typically made at 10 to 50 L scale, but many stem cell and stem cell–derived drug products are targeting larger production capacity. The manufacturing scale is primarily triggered by patient dosing requirements and patient population size.
Cell dosing requirements for mesenchymal stem cell (MSC) or pluripotent stem cell (PSC) therapies are estimated to need up to 109 cells/patient, with some indications having anticipated market sizes needing hundreds of thousands of doses. It is anticipated that some commercial allogeneic stem cell manufacturing will require batch sizes in the 200 to 2,000 L range, producing 1011–1014 cells/year for a single product to reach commercial viability.3
The Start
The key to designing a stem cell or stem cell–derived product manufacturing facility is to address the multivariable problem of constraints, goals, adjacencies, and cost. The facility must be designed and built to promote simplified production by the operators, consider workflows, enable safe operation, and ease regulatory compliance. All manufacturing flow paths through the facility must be studied, including product, personnel, raw materials, waste, and equipment. Too much focus on the primary process can lead to insufficient study of support functions such as media preparation and delivery, kitting, quality control (QC) laboratories, or warehousing. Taking a broader view of the facility will allow for a more comprehensive design.
Open vs. Closed
One of the biggest considerations when designing any biomanufacturing facility is whether the process is “open,” “closed,” or a mix of the two. Cells in an open process are exposed to the surrounding environment and the operators. Closed processes are designed such that the surrounding environment and operators are not exposed to the process or vice versa. This is accomplished using stainless steel vessels and piping, single-use systems and tubing assemblies, isolators, and other means.
Open processes have an inherently greater risk of contamination than closed processes and require a higher-grade environment and a more conservative facility design; thus, for streamlined facility design and optimum operator and product safety, closed processes should be considered wherever possible. Although parts of a process—such as a single-use bioreactor for cell expansion—may be closed, all processing steps—such as inoculation or harvest—must be considered during facility design. A block flow diagram (see Figure 1) helps identify each process step and assign an open or closed label. If a processing step is not currently closed, the means for “closing” it should be considered.
It is important to understand that stem cell products are living cells that cannot be sterilized by filtration, heat, or any other current method. Therefore, full aseptic processing is required from start to finish. There must be a focus on maintaining aseptic techniques to prevent the introduction, transmission, or spread of communicable disease from the outset of operations.4 Because many stem cell processes start their development in academia or small research laboratories, there is a trend toward processes using many manual open steps to limit initial costs and ease operations. As these processes graduate to larger clinical trials or commercial manufacturing, early process development should focus on using closed processing and more automated procedures to lower the risks of contamination and improve efficiency.
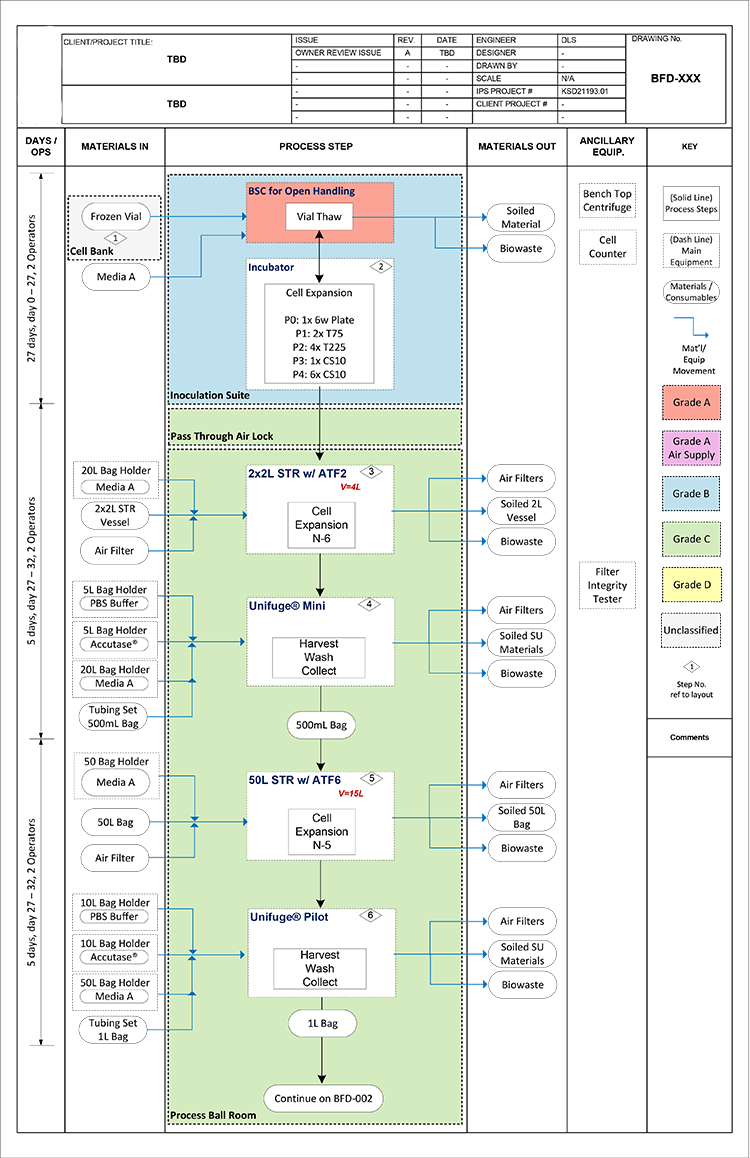
Product development efforts should keep scalability in mind early during technology transfer implementation to ease this transition. By minimizing the amount of open processing required in the early stages of manufacturing, there is an opportunity for substantial savings in cost and footprint in both equipment and building utilities. Occasionally, a specific step in an otherwise closed process cannot be closed due to project timeline, cost, or other reason. In this scenario, a dedicated room and additional transition airlocks are likely required to segregate the open process. Open processing requires, at minimum, working in a biosafety cabinet (BSC) with a Grade A air supply in a Grade B room background.4
Risk Assessment
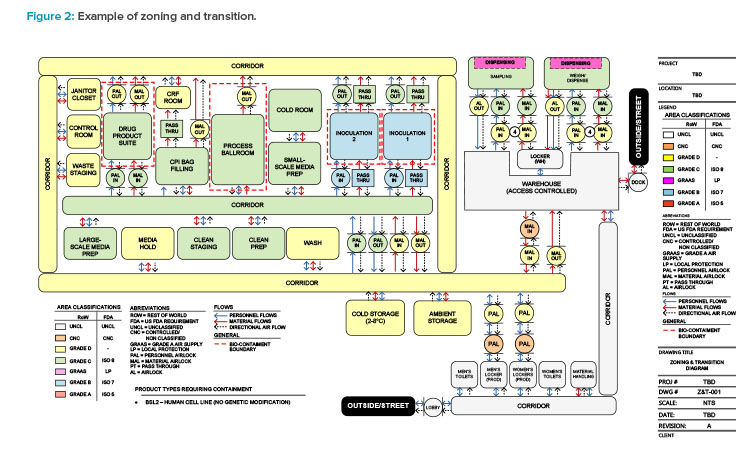
Before a facility layout is developed, a set of preliminary risk assessments should be performed. Risk assessments relate to business, quality, and safety functions of an organization and are a requirement to meet Centers for Disease Control and Prevention (CDC) and National Institutes of Health (NIH) guidelines.5 Specific risk assessments can be performed for product contamination risk, process reliability (product loss), product cross-contamination risk (product to product, batch to batch), and environmental, health, and safety (EHS) risk. Zoning and transition diagrams (see Figure 2) are a great tool to understand potential adjacencies of different process manufacturing operations and can be used to facilitate a risk assessment. The risk assessment typically falls into one of the following categories.
Single-Product Facilities
The risk profile for single-product facilities is relatively low compared to other types of facilities. The focus of the assessment is typically on aseptic operation, batch segregation, and worker safety. Based on risk assessment, these facilities may be designed with bidirectional flows with proper airlock design for required material and personnel transitioning. Single-product facility designs often allow for consolidating multiple closed functions into a single room.
Multiproduct Facilities
Potential for cross-contamination increases in a multiproduct facility, so both facility design elements and operational elements must be considered. Individual products must have a designation and secure manufacturing sequence. Several means can be leveraged to reduce the risk profile, including campaigning, physical segregation, once-through heating, ventilation, and air conditioning (HVAC) design, and careful sanitization. ISPE Baseline® Guide Vol 7: Risk-Based Manufacture of Pharmaceutical Products6 outlines a scientific risk-based approach for managing the risk of cross-contamination within multiproduct facilities.
Contract Manufacturing Organizations (CMOs)
CMOs often have special considerations due to the nature of their business, which involves insourcing other organizations’ manufacturing operations. CMOs often have multiple clients, processes, and products housed under the same roof, and the multiproduct risks are magnified. This increased risk often becomes a concern for their clients and frequently drives a compartmentalized suite approach for phase II clinical trial manufacturing through commercial-scale manufacturing.
Regulatory Compliance
Too often, it happens that the initial layout and concept designs are performed without fully considering the relevant regulations and guidelines. First, the markets in which the product will be sold must be defined. This will drive which good manufacturing practices (GMPs) the facility design must follow. Next, the location where the facility will be constructed must be established. This location will drive things like biosafety requirements, building code, and environmental and waste handling requirements. The following are some of the key regulations that constrain facility design and provide insight into the regulatory expectations.
United States
The following GMP regulations from the Code of Federal Regulations are relevant:
- 21 CFR Part 1271—Human Cells, Tissues, and Cellular and Tissue-Based Products4
- 21 CFR Part 210—Current Good Manufacturing Practice in Manufacturing, Processing, Packing, or Holding of Drugs; General7
- 21 CFR Part 211—Current Good Manufacturing Practice for Finished Pharmaceuticals8
- 21 CFR Part 600—Biologics Products: General9
- 21 CFR Part 610—General Biological Products Standards10
- 21 CFR Part 11—Electronic Records; Electronics Signatures11
The following GMP guidelines from the US Food and Drug Administration should be considered:
- “Guidance for Industry. Current Good Tissue Practice (CGTP) and Additional Requirements for Manufacturers of Human Cells, Tissues, and Cellular and Tissue-Based Products (HCT/Ps).” December 201112
- “Guidance for Industry. Sterile Drug Products Produced by Aseptic Processing—Current Good Manufacturing Practice.” September 200413
The following safety regulations are relevant:
- 42 CFR 73—Select Agents and Toxins Regulations14
- OSHA regulation, 29 CFR 1910.1030—Bloodborne Pathogens15
The following safety guidelines should be considered:
- CDC/NIH—Biosafety in Microbiological and Biomedical Laboratories (BMBL) 6th ed.5
- NIH Guidelines for Research Involving Recombinant or Synthetic Nucleic Acid Molecules16
- Small-Scale < 10 liters – Appendix G
- Large-Scale > 10 liters – Appendix K (includes manufacturing)
Finally, the following federal, state, and local building codes, as well as other specific guidelines, must be addressed:
- International Code Council (ICC)
- National Electrical Code (NEC)
- Occupational Safety and Health Administration (OSHA)
- National Institute for Occupational Safety and Health (NIOSH)
- National Fire Protection Association (NFPA)
- Americans with Disabilities Act (ADA)
Europe
The following GMP regulations are relevant:
- Regulation (EC) No 1394/2007 of the European Parliament and of the Council of 13 November 2007 on Advanced Therapy Medicinal Products and Amending Directive 2001/83/EC and Regulation (EC) no 726/200417
- European Commission. “EudraLex, Volume 4: Good Manufacturing Practice—Guidelines on Good Manufacturing Practice Specific to Advanced Therapy Medicinal Products.” 18
- European Commission. “EudraLex, Volume 4: EU Guidelines for GMPs for Medicinal Products for Human and Veterinary Use. Annex 1: Manufacture of Sterile Medicinal Products.” 19
The following biosafety regulations and guidelines are relevant:
- Directive 2009/41/EC of the European Parliament and of the Council of 6 May 2009 on the Contained Use of Genetically Modified Micro-Organisms20
- Specific regulations and guidelines issued by EU member states
For building codes, specific regulations and guidelines issued by EU member states should be followed.
Rest of the World
Many countries, such as Canada, Brazil, China, and Mexico, have country-specific regulations and guidelines. Building codes are often country-specific.
The following GMP and biosafety regulations and guidelines may be relevant:
- Pharmaceutical Inspection Convention/Pharmaceutical Inspection Co-Operation Scheme (PIC/S) PE-009-16 Annex 2A—Manufacture of Advanced Therapy Medicinal Products for Human Use21
- The World Health Organization (WHO) generally publishes GMP and biosafety guidance for countries that do not issue their own regulations or guidelines
Considerations
Although not all the regulations listed are relevant for every facility, the engineers and architects designing the facility need to understand which aspects are critical. The recent update to EU GMP Annex 1 (2022)19 has added the requirement for facilities to have a contamination control strategy (CCS). While regulators have always expected facilities to have a documented plan to control contamination, existing approaches may not always be coordinated between different departments (e.g., QC, quality assurance, or manufacturing), leading to disjointed data from original qualifications, validations, process controls, and environmental monitoring.
Additionally, corrective and preventive actions taken in response to deviations and trend analyses may lack both integration into a comprehensive strategy and a link between critical control points and evaluations of control effectiveness (design, procedures, technology, and organization).22 However, a holistic view is proposed in Annex 1 (2022)19 for particulates, microbial, and pyrogen contamination. There is still discussion and debate around exactly how CCSs should be developed and documented. With that said, the ECA Task Force on Contamination Control Strategy has created the guideline “How to Develop and Document a Contamination Control Strategy,” which should serve as an excellent starting point.22
| Category | Time | Biosafety Cabinet | Isolator | Comments |
|---|---|---|---|---|
| Capital Costs | ||||
| Capital Facility | Initial cost | $ 533,600 | $ 413,000 | Estimated $/sq ft |
| Initial cost | $ 119,571 | $ 1,117,550 | From equipment quotes | |
| Initial cost | $ 8,500 | $ 78,500 | Soft cost allowance - 7% of capital | |
| Initial cost | $ 8,500 | $ 316,650 | Soft cost allowance - 7% of capital + vendor-supplied commissioning costs |
|
| Maintenance Costs | ||||
| Maintenance | Annual | $ 14,500 | $ 67,500 | Soft cost % of capital |
| Labor Costs | ||||
| Manufacturing Operators | Annual | $ 300,000 | $ 300,000 | $150,000 per full-time equivalent operator (2 operators) |
| Cleaning Labor | Annual | $ 150,000 | $ 75,000 | $150,000 per full-time equivalent operator (2 and 1 operators) |
| Operational Costs | ||||
| Heating, Ventilation, and Air Conditioning | Annual | $ 136,207 | $ 37,196 | |
| Environmental Monitoring | Annual | $ 150,000 | $ 75,000 | Assumed value |
| Gowning Materials (Grade B) | Annual | $ 103,181 | $ - | |
| Gowning Materials (Grade C) | Annual | $ 93,683 | $ 93,683 | |
Operational Challenges
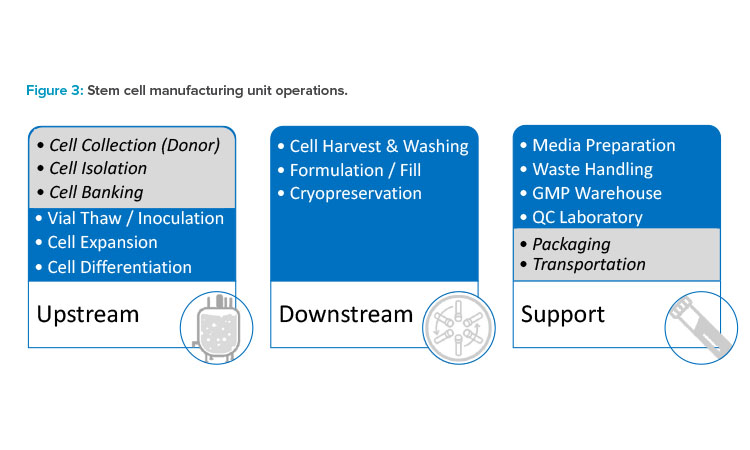
To better understand the challenges posed by large-scale stem cell and stem cell–derived product manufacturing, it is easiest to review the operations found in most stem cell processes and facilities (see Figure 3). The list of considerations is certainly not all-encompassing, but it should provide a strong overview of the major challenges. The manufacturing of stem cells begins with donor selection, but this article focuses on the manufacturing facility starting with inoculation. Cell line development—including cell collection, cell isolation, cell activation, or cell banking—is considered outside of the scope of this article.
Inoculation
In most applications, initial vial preparation requires manual and open processing, necessitating a Grade A environment. This environment can be achieved in a couple of ways, either by using BSCs with a Grade B background or within an isolator in a Grade C or Grade D background, depending on the regulatory environment (EMA vs. FDA). Although isolators provide the most qualifiable sterile environment, the choice between the two methods usually falls into a debate over cost, schedule, operational flexibility, and risk associated with batch loss.
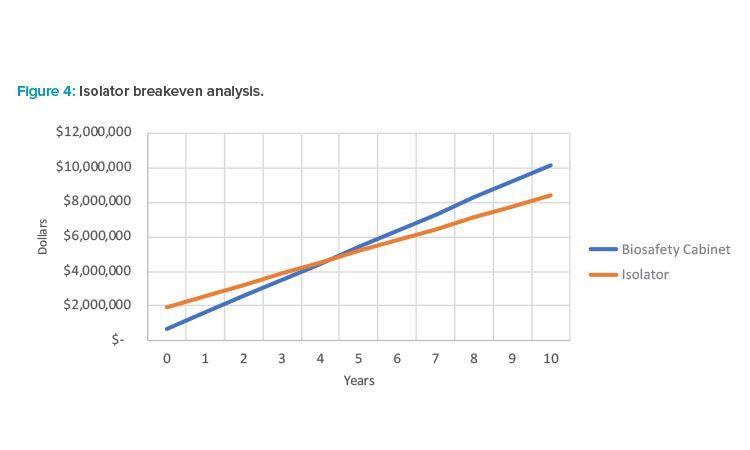
At the time of writing, the most common choice is to perform inoculation in a BSC with a Grade B background. BSCs have the benefit of process versatility, whereas isolators are better suited for mature processes due to the need for customization. Often overlooked is the fact that Grade B space can become very expensive in both capital and operation costs when architectural finishes, HVAC, gowning, cleaning, and turnover time are considered. Some studies (see Table 1 and Figure 4) have shown that isolators, while often having a higher capital cost, can have a relatively short breakeven period when considering costs over the lifetime of the equipment. Ultimately, this should be investigated on a case-by-case basis when designing a facility.
Cell Expansion
Selecting the right scalable platform for stem cell expansion can greatly affect facility design. Although there are technologies available that can be adopted for stem cell manufacturing from existing bioprocessing standards, the specific requirements for stem cell processes should be carefully reviewed and addressed before using existing solutions. Many stem cell lines are being developed using traditional adherent plate flatware, such as T-flasks and multilayer cell culture vessels. While some autologous or small-scale allogeneic processes may use flatware for commercial production, this is not feasible on a large scale due to the operational challenges of increased equipment size and complex automation.
Alternatively, many of the commercially available closed adherent bioreactors have been developed with gene therapy or other traditional biologic products in mind, where the products are expressed extra-cellularly, or the cells are lysed to recover intracellular products. Harvest of live cells poses a major challenge in many of these adherent platforms due to the type of substrate that is used as an anchor for the cells. There are some commercially available adherent platforms with substrates designed to allow for high yield and viability while harvesting live cells, but these platforms are not yet available in the capacity required for large-scale manufacturing. With that said, these platforms could serve as a useful seed train design solution to close the expansion operations as early as possible in the process.
Given the current technology, suspension-based approaches for adherent stem cells, typically coupled with perfusion technology for continuous media exchange, have proven to be more scalable. The two current methods are microcarrier-based suspension and cell aggregate suspension. Microcarrier suspension uses micro-porous beads or other media that allow adherent cell lines to attach and grow in high density while suspended within a liquid medium.
This approach allows the use of commercially available and proven scalable suspension bioreactors but introduces an additional process operation. To harvest cells that are adhered to microcarriers, the cells must first be dissociated from the beads. This is typically performed with the addition of a dis-sociative agent, such as trypsin. After the disassociation, the beads then also need to be separated from the cells. This is typically done via some form of filtration. Alternatively, aggregate suspensions are an acceptable growth method for some stem cell lines, wherein groups of cells form colonies or aggregates to support growth while in suspension.
Monitoring and controlling the cell density and aggregate formation is key in these processes to maximize cell growth and viability. In addition, cells will periodically require disassociation methods between interim growth expansion steps to allow for fresh aggregate formations in new media or transfer into final product formulation.
Stem cell culture processes and stem cell line development are not yet mature; thus, process development to support an increased number of stem cell passages poses many challenges. Given this, options for reaching large-scale production may be limited. Facility designs will likely need to have consideration for both scaling up and scaling out to reach production targets. If developing a stem cell process for a single large production bioreactor is not possible, the facility may need to support a process that has multiple smaller reactors.
Cell Harvest and Washing
A key area where stem cell manufacturing differs from traditional biotech is harvest and purification. Traditional biotech may use a combination of centrifugation, depth filtration, chromatography, tangential flow filtration (TFF), or other forms of filtration to remove cell debris or target a specific protein, whereas stem cell harvesting typically has fewer unit operations. The target of stem cell harvesting is to collect individual live cells. This can be completed by a series of operations, including dissociation (if necessary), quenching, washing, concentration, and collection.
As a cell product, every harvest step requires a focus on critical quality attributes such as cell concentration, total cell count, and cell viability. The cells themselves must be maintained in good condition throughout the operation, limiting equipment choices for wash and concentrate operations where there is potential for high shear forces or other damaging impacts to the cell product. To choose the correct operating methodology and equipment, each process step requiring the manipulation of the product volume and composition must be closely investigated to find the most appropriate solution.
Beyond the harvest process, other operations may require adjustments to cell concentration. Many stem cell expansion processes require regular media exchange to replenish nutrients and remove cell waste byproducts. This media exchange can be done continuously (perfusion) or via discrete media exchange where the working volume in a bioreactor is reduced and a bolus of media is added. This type of media exchange can improve cell density and overall health.
Stem cell–derived products may require many different specialized media types to be added discretely. This discrete media exchange can be necessary to direct cell differentiation into the desired pathway. Each of these steps necessitates the removal of spent media and the introduction of new media to the product liquid without negatively impacting the product cells. The choice of technologies that results in maximum processing rates and volumes can greatly benefit the overall manufacture of these cell products.
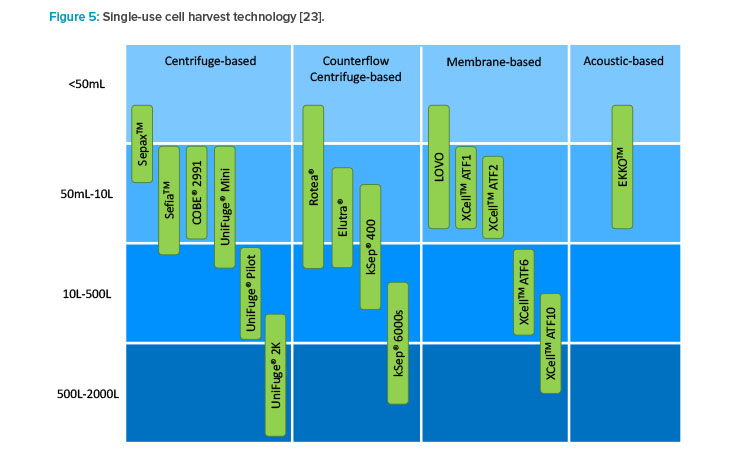
There are several technology platforms used for cell harvest or media exchange, but not all can meet the needs of large-scale manufacturing (see Figure 5). Single-use equipment options are the leading solution to this design challenge and include centrifugation, acoustic wave separation, TFF, and alternating tangential flow (ATF) filtration (see Figure 4). Acoustic wave separation is an interesting and promising technology, but it is not commercially available for large-scale manufacturing.
Although TFF or ATF may be appropriate for continuous media exchange, the throughput of these systems is typically too slow for discrete media exchange or harvest operations. Presently, single-use centrifugation is the most effective solution for cell harvest or media exchange. It allows for dissociation, quench, wash, concentration, collection, and discrete media exchange all with the same piece of equipment with relatively short processing times. Designing processes to use the same piece of equipment for multiple unit operations can allow for a smaller facility footprint, higher equipment utilization, and reduced capital cost.
Formulation, Filling, and Cryopreservation
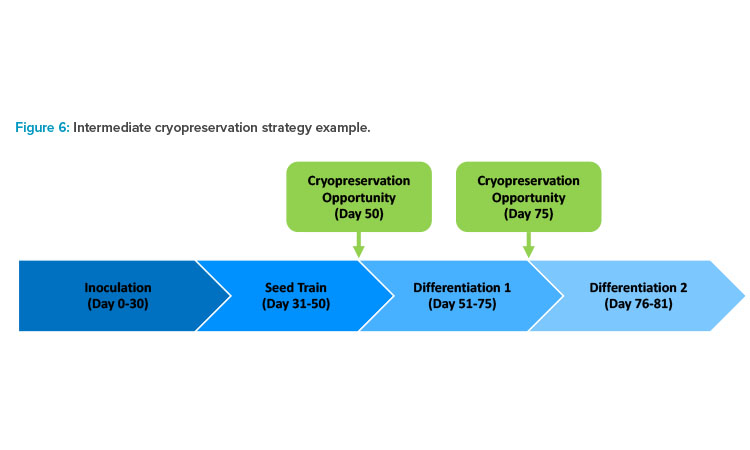
Some large-scale stem cell processes require multiple types of filling technologies and formats for intermediate and final products. Given the long timelines for cell expansion and differentiation at large-scale, it can be advantageous to strategically decouple operations by cryopreserving and storing intermediate products. Decoupling operations such as seed trains and production bioreactors can reduce the impact of a batch failure and ease batch scheduling.
Consider, for example, a process that has a failure during the differentiation phase. If that process has a cryopreservation operation directly before differentiation, differentiation could be restarted immediately using the cryopreserved intermediate, losing only a week or two of productivity (see Figure 6). A process that does not have an intermediate cryopreservation step would have to start from a vial from the primary cell bank, losing several weeks or months of productivity.
Filling and cryopreservation of most cell therapy products is a small-scale operation. The cells are formulated with a cryoprotective agent, such as 10% dimethyl sulfoxide (DMSO), to prevent the formation of crystals during freezing. The formulated product is then filled into a small number of cryovials or cryobags. The cryoprotective agent can affect cell viability if kept in a liquid state for extended periods, so the formulation step starts a “freeze window” that is typically one to two hours. The cryovials or cryobags are then transported to a benchtop-controlled rate freezer where the vials or bags are brought down to the target temperature using liquid nitrogen. This operation is adequate for most existing cell therapy manufacturing processes, but large-scale stem cell therapy processes require large-scale solutions.
To improve logistics and material handling, it can be advantageous to fill and freeze intermediate products in larger formats than traditionally done in cell therapies (i.e., 500 mL and larger). Consider an instance where a differentiation bioreactor requires 4 L of cryopreserved intermediate to inoculate. It is far easier for operators to handle four 1-L bags, or even one 4-L bag, during the thawing and inoculation process than twenty 200-mL bags. This same logistic and material handling challenge is encountered during freezing. It is much easier to move a smaller number of larger bags from a filling operation to a freezing operation than it is to move a large number of small bags.
Most automated technologies available for filling bags in the 200 mL to 5 L size range are isolated high-speed units designed for intravenous (IV) fluids. These isolated high-speed filling lines are designed to fill at a minimum of 500–1,000 bags/hr. This capacity would be excessive for most stem cell therapy products and comes with a high capital cost and large footprint. Another option to a traditional high-speed bag filling line is a closed, single-use, manifold-style filler.
A growing number of vendors are tailoring their filling products to cell therapy processes, and there are now a handful of vendors that have sin-gle-use manifold-style offerings fit for 50 mL to 20 L bags. One of the challenges of using a larger bag format is that there is little to no publicly available data on post-freeze cell viability. In the short term, manufacturers will have to generate their own experimental data to prove the large-scale technology is capable of freezing intermediate products without large cell loss.
Media Preparation and Delivery
Media is an essential component of all cell culture processes, but supplying media to large-scale stem cell processes can pose some unique challenges. These challenges can come from both the stem cell process and the composition of the media itself. Some of the major challenges posed by the process include sterile preparation, large process scale, continuous perfusion requirements, bolus media additions, and preparation and delivery of several types of growth and differentiation mediums.
Because the final cellular product cannot be filtered in a sterile fashion, the entire process, including media production, must be performed in a sterile (i.e., not bioburden-controlled) fashion. This means additional filtration and testing is required compared to a traditional biomanufacturing process and could drive the need for larger or dedicated QC laboratories. Small-scale cell therapy processes can leverage external suppliers to provide pre-prepared sterile liquid media in single-use containers.
The sheer volume of media required for large-scale stem cell processes makes purchasing preprepared media impractical and thus onsite preparation from powdered media stock is necessary. Stem cells are notoriously media hungry, and some stem cell processes require continuous perfusion of media to sustain the cells. Continuous perfusion could mean an entire reactor volume is turned over every day of the process. Cell expansion and differentiation processes can both last several weeks, potentially requiring more than 10,000 L of media to complete a single 200 L production reactor drug sub-stance batch.
| Storage Type | Ambient | 2°C–8°C | –20°C | –190°C |
|---|---|---|---|---|
| Storage Basis | 6 months | 4 months | 12 months | 12 months |
| Storage Unit | Pallet | Pallet | 23 ft3 freezer | 750 L liquid nitrogen freezer |
| Quantity | 700 | 120 | 10 | 12 |
Delivery of media from the preparation equipment to the bio-reactors can pose additional challenges, making the development of a robust media delivery strategy essential. One of the biggest decisions is whether single-use or stainless steel media preparation and delivery equipment will be used or whether a combination of the two is required. One strategy to consider is using single-use media preparation and delivery equipment for the lower volume seed train while using stainless steel media preparation and delivery equipment for the larger product or differentiation bioreactors.
A typical media system would include a media preparation tank or single-use mixer, a sterile filtration skid or tubing set, a media hold tank or sin-gle-use tote, and a distribution system or tubing set. An analysis of capital and operating costs for single-use vs. stainless steel equipment can help inform the development of the media strategy. The chemical stability of the media, especially the chemical stability of any growth factors or small molecules, could drive the need for cold storage or limit the window between preparation and expiration. The need for a bolus addition of media to a bioreactor when switching between differentiation media types may mean cold media (2°C–8°C) needs to be warmed to 37°C before it is added to the reactor.
Unique strategies and schedules may have to be developed for processes that use multiple production or differentiation bioreactors. Take, for example, a process that uses four 500-L differentiation bioreactors. Each bioreactor could have a dedicated set of media equipment, or a larger batch of media could be prepared, held, and distributed to all the bioreactors. If cold storage and bolus media additions are required, then heat exchangers need to be added as part of the distribution to warm the media before delivery to the bioreactors.
Biowaste Handling
Human cell lines are considered to be potentially infectious and within the scope of the OSHA Bloodborne Pathogens Standard (BPS) unless the specific cell line has been characterized to be free of recognized bloodborne pathogens.24 Additionally, the BMBL recommends that human cells should be handled using Biosafety Level 2 (BSL-2) practices and containment.16 These biosafety implications are important when it comes to handling waste because waste handling procedures must adhere to the results of a site-specific risk assessment. The following recommendations are typical and assume the only biohazards are human stem cell products without genetic modification.
Solid Waste
All contaminated solid waste from the production area shall be decontaminated in accordance with applicable local, state, and federal regulations. This decontamination can be performed onsite or offsite as long as it is packaged correctly for transport.16
Waste Flows
Designated waste flows should be considered for contaminated waste within the facility layout to avoid any source of cross-contamination. As a best practice, waste flows should be unidirectional and segregated from other material flows wherever possible. Where waste flows cannot be segregated physically, they should be segregated by time. Once the decontamination is complete, the waste can be moved to a central treatment area where a combination autoclave/shredder unit can be used to treat the material, which can then be disposed of as municipal solid waste. Alternatively, if bio-waste hauling services are available at the site, the biowaste cart can be moved to a waste storage area for offsite treatment.
Liquid Biowaste
Large volumes of liquid biowaste generated in Grade C or Grade D cleanrooms should be collected via a dedicated, fully welded biowaste drain system in the GMP rooms. If the drain system is located underground, double-contained piping with active leak detection should be considered. Although only required as part of BSL-3 design, as a best practice, the drain system should use closed connections/covers and drain vents that are outfitted with 0.2 µm or HEPA-grade filters to maintain a closed boundary and prevent the release of any airborne organisms. The waste can then be treated via a qualified decontamination method. This can be performed either chemically or thermally. The thermal option is a more conservative and flexible approach, and two system designs are typical: dual batch (using two alternating tanks for batch operations) or continuous decontamination.
The impact of the biological oxygen demand (BOD) of the liquid waste streams on the site waste permit should also be considered. For large-scale stem cell manufacturing facilities that use perfusion or regular discrete media exchange, large volumes of barely spent media (i.e., high-BOD waste) could be designated for the sanitary sewer. The BOD content of the waste should be analyzed and compared with the site permit limits. If necessary, the facility may require a means for segregating and collecting the high-BOD stream for hauling by tanker truck and treatment offsite.
GMP Warehouse and Material Staging
Large-scale stem cell manufacturing facilities require significant and diverse storage space. Large amounts of short-term storage and laydown spaces are required in the clean core for staging single-use materials. For the GMP warehouse, ambient storage is required for single-use components and other shelf-stable materials. Storage at 2°C–8°C and –20°C is required for powdered media and other raw materials, respectively. Cold rooms would likely be required to support the amount of 2°C–8°C storage required, whereas –20°C storage may be able to be supported by upright freezer units. Cryogenic (–180°C) dry-phase nitrogen dewar storage is required for cryopreserved intermediate material. Table 2 displays an example of GMP warehouse storage requirements for a large-scale stem cell therapy facility.
Quality Control Laboratory
Large-scale stem cell manufacturing facilities require significant analytical support. The extensive amount of process sterility testing necessitates QC laboratory space to be located with access from the clean core for ease of sample delivery. QC laboratories also face the same biowaste handling requirements as the manufacturing area.
Although some stem cell drug products may be cryo-preserved, others may need to be delivered to the clinic fresh (i.e., 2°C–8°C). The latter scenario requires very tight timelines from completion of filling to delivery to the clinic, similar to an autologous facility and product supply chain. Tighter product delivery timelines lead to ultra-fast release testing and drive the need for increased in-house capabilities. The QC area design should consider the ability to receive and test samples for the following:
- Product
- Media sterility/bioburden
- Raw materials
- Facility utilities monitoring
- Environmental monitoring
- Aseptic process simulation
- Revalidation — clean-in-place (CIP)/sterilization-in-place (SIP)/cleanroom
Additionally, space requirements in the QC area for equipment to support the following assays should be considered:
- Bioburden and sterility – endotoxin (LAL), mycoplasma (PCR), BACT/ALERT, Gram stain
- Biological indicators – growth
- Markers/purity/potency – nuclear/cytoplasmic markers, incubation, ELISA immunocytochemistry, high content imagery
- Karyology – G-banding/cytogenetic
- Content – hemocytometer, total count, viability
- General – appearance, conductivity, osmolality, pH
- Total organic carbon (TOC) – rinse water and swabs
- Environmental monitoring – total particulate, nonviable particulate, airborne viable particulate, surface viable particulate
- Gases and raw materials – identity
Conclusion
As single-use process equipment continues to evolve, there will be new opportunities to apply principles from the existing bioprocessing industry to the large-scale cell therapy field. Designing these facilities is a complex system of challenges based on safety, cost, and efficiency that requires leveraging constantly evolving technology. Doing so while meeting the needs of regulators, manufacturing, QC, facilities, and business units requires a team with the right expertise and experience.
About the Authors




