Today’s Pharma and Biotech Projects: A Phased Approach
This article revisits the concept of phased engineering, procurement, and construction (EPC) and updates it with risk-based considerations specifically regarding the commissioning, qualification, and validation (CQV) of general life-cycle principles for pharma and biotech projects. Enhancing the relationship between phases of a project, advanced planning, and more formal management of uncertainties could maximize the overall efficiency, productivity, and effectiveness of engineering projects. The use of a risk-based phased EPC-CQV approach will be key in meeting the increased global demand for capacity and successful emergency preparedness efforts. This article revisits an article on this topic published in Pharmaceutical Engineering® in 1997.
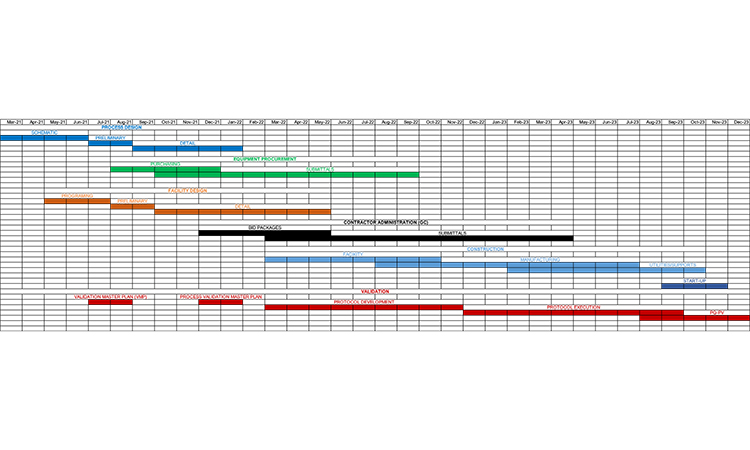
The general outline of a pharma or biotech engineering project includes phases and tasks that typically span 2–3 years (Figure 1). The recent pandemic emergency response and the level of readiness demonstrated by reusing/converting existing capacity have shown that compression of these project timelines can be achieved in practice.
In today’s business environment, where every project is a one-shot approach with aggressive objectives (higher quality, shorter schedule, diminishing resources), there is a tendency to compress a project to the point where many of the normal development activities (e.g., process definition, facility design, procurement, construction) take place at once. We used the phased approach (Figure 1) to compress the schedule. That can be supported by risk-based management to select and prioritize critical project aspects. Concurrent activities to compressed project activities may offer some short-term relief, but this approach can cause coordination problems within the design and interference problems during construction. This will lead to cost overruns and schedule delays due to rework of the design or the need to make changes in the field.
This article is not intended to be an in-depth presentation of all aspects of project design, construction, commissioning, qualification, and process performance qualification and validation (PPQ/PV). Instead, the article discusses phased EPC-CQV, the relationship between the phases of a project, and how advance planning could maximize productivity.
Challenges in Today’s Projects
The current global pandemic has placed considerable pressure on biopharmaceutical manufacturer suppliers to meet increased demands, and various pharma manufacturers are working toward specific goals. Individuals involved in specific pharmaceutical manufacturing segments want the most flexible process and facility design possible while still incorporating the best technology and anticipating future industry trends. Quality assurance is involved at earlier stages, imposing requirements for regulatory compliance and validation, which have changed significantly with the introduction of risk-based CQV and the ongoing use of life-cycle validation (continuous process verification [CPV]) instead of annual quality reviews. Financial planning requires a higher return on investment, and capital funds appropriation.
Under these conditions, there is a tendency to use the “shotgun” method to finish the project by attempting to run as many concurrent project tasks as possible. Without a higher level of coordination that uses risk management and risk-based decision-making to manage conflicting scenarios and the impact of uncertainties, the shotgun approach is only partially effective. This is because work is completed without the required input, resulting in engineering and construction rework that adds to project costs. Engineering rework resulting from untimely input or poor project coordination can add 10% or more to the cost of a project depending on the circumstances. Changes to a project during construction can add 15%–25% to a project cost. Negative schedule impacts can include late changes that can end up extending the project schedule.
The optimal situation is to understand the relationship between different aspects of a project, to overlap the phases as much as is practical, and to ensure that the proper inputs occur so that rework is not required. The planning and design of a project generally follow the development of the process or manufacturing area, along with procurement of major equipment and the design of the corresponding facility. Once the design is well underway, construction management and construction will commence. At the completion of the construction phase, commissioning and the field phase of qualification will take place. The exact relationship between the activities may vary from project to project along with their relative durations and the degree of overlap.
Validation and Life-Cycle Management
Validation as defined by the GMP regulations in the largest International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) regions has changed significantly since 2011, which is when the US FDA introduced the use of explicit risk-based life-cycle management considerations and ongoing, continued, or continuous process validation as a new way to assess product quality consistency, process robustness, and facility compliance.2
The FDA defines process validation as the collection and evaluation of data originating from three stages:2
- Stage 1: Process Design/Characterization: The commercial manufacturing process design is defined based on knowledge gained through development and scale-up activities.
- Stage 2: System Qualification/Continued Process Performance Qualification: The process design is evaluated to determine if the process is capable of reproducible commercial manufacturing.
- Stage 3: Continued Process Verification: Ongoing assurance is gained during routine production where the process remains in a state of control.
This approach is also endorsed by the European Medicines Agency (EMA). Annex 15 of the EU’s GMP regulations issued in 2015 states that for products developed under a quality by design (QbD) framework, a risk-based process control strategy should be scientifically established during development and tested in the PPQ qualification stage to provide a high degree of product quality assurance and, most importantly, safety and efficacy.3 This strategy should be applied to evaluate the required attributes for incoming materials, critical quality attributes, and critical process parameters to confirm product realization and regularly evaluated based on the best knowledge and data available.
| Qualification | Definition |
|---|---|
| User requirements specification (URS) | The set of owner, user, and engineering requirements necessary and sufficient to create a feasible design meeting the intended purpose of the system. |
| Design qualification (DQ) | The documented verification that the proposed design of the facilities, systems, and equipment is suitable for the intended purpose. |
| Installation qualification (IQ) | The documented verification that the facilities, systems, and equipment, as installed or modified, comply with the approved design and the manufacturer’s recommendations. |
| Operational qualification (OQ) | The documented verification that the facilities, systems, and equipment, as installed or modified, perform as intended throughout the anticipated operating ranges. |
| Performance qualification (PQ) | The documented verification that the systems and equipment can perform effectively and reproducibly based on the approved process method and product specification. |
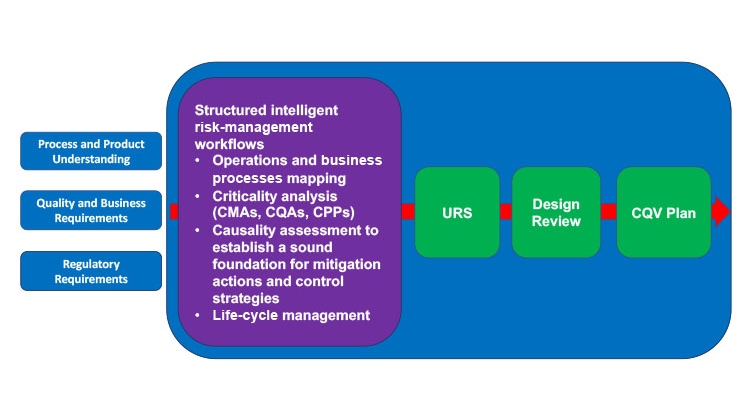
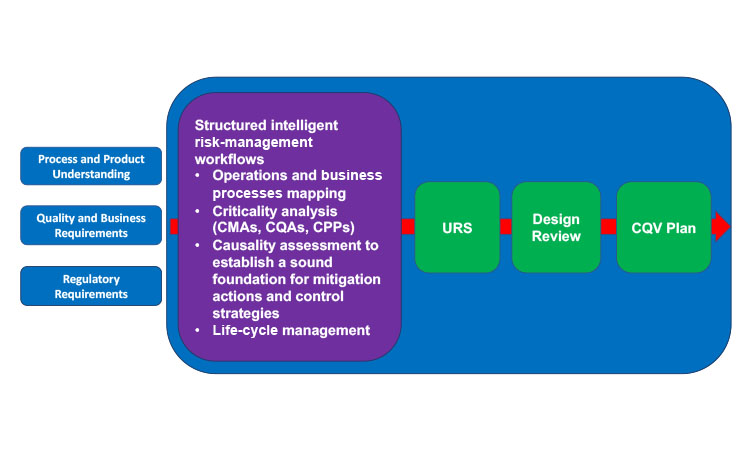
Both the FDA and EMA guidelines emphasize the importance of applying risk-based procedures to justify and manage all critical quality aspects over the life cycle of a project, from CQV to control strategy definition to postapproval changes that may occur on the process, plus drug product formulation, facilities, methods, suppliers, and supply chains. CQV activities are heavily influenced by the way a process is designed and the depth of knowledge available (Figure 2). The qualification stage of an existing process design should use risk management not only to be comprehensive and to help close any knowledge gaps, but also to help prioritize activities, define a suitable level of effort in qualification, and document activities capturing the context and rationale used (Table 1). Thus, risk management has been considered a new GMP requirement since 2015.
Efficient and successful process validation depends on the use of a structured workflow that includes operations or business processes mapping, criticality analysis, risk evaluation, risk-benefit balancing, threshold definition (between acceptable/residual risks and unacceptable risks that require mitigation), adequate controls, a life-cycle management plan for recurrence of risk reviews, and a mechanism to incorporate continuous improvement opportunities (current project and across the portfolio).
Digital Risk Management
The adoption of digital risk management solutions to speed up the introduction of risk-based CQV and modern risk-based life-cycle management has been gaining momentum. The use of digital solutions holds great promise as it establishes a robust knowledge management framework on which to build consistency and traceability over a project and across multiple projects.4
There is continued support from the FDA regarding the general use of automation and digital solutions in pharma processes. The FDA is expected to release a new guidance document, Computer Software Assurance for Manufacturing, Operations and Quality System Software, in 2021.5 Computer software assurance (CSA) aims to bring agility to the adoption of new digital platforms supporting manufacturing operations and will extend the existing computer systems validation (CSV) requirements by enforcing verification over life cycle in a CPV sense.
Conditions exist today that promote an enhanced approach to process design and qualification activities that will result in acceleration and agility in postapproval change management over the project life cycle, as described in ICH Q12.6 These processes take advantage of state-of-the-art digital tools to automate risk-based CQV and CPV programs. The use of structured risk and knowledge management approaches will promote faster delivery of safe and high-quality products to the patients that need them.
Validation Master Plan
Development of a pharmaceutical project is not complete without consideration of the process, equipment, and facility validation. Today, planning of the validation program begins as soon as the process and facility concepts are firm. Conceptual flow diagrams and equipment/facility layouts are reviewed to ensure that the proposed design can be validated prior to operation. Validation planning also establishes acceptance criteria for the various equipment and process systems, a key part of the specification and procurement of these systems.
I. Overview – Why a master plan?
|
VI. Automation concepts and objectives
|
II. Validation activities
|
VII. Validation resources
|
III. Facility
|
VIII. Documentation schedule outline
|
IV. Process description drug substance
|
IX. Validation execution schedule
|
V. Process description drug product
|
X. Other activities
|
This early development approach avoids later changes during design or construction and anticipates the time (schedule) and resource (personnel, training, cost) demands that will be placed on the team during final execution of the commissioning and qualification (C&Q) and validation in the field. The validation master plan is prepared to outline the overall plan for the project C&Q and validation in terms of what systems will be commissioned, qualified, and validated and to what extent. It also identifies a tentative schedule, personnel requirements, and training resources (Table 2). The master plan is also a good vehicle to use to begin discussions with the regulatory agencies regarding their participation in the review and approval of the process and facility.
Commissioning and Start-up
Once construction is completed, the commissioning and start-up phase of the project can begin. This phase includes the following components:
- Testing by equipment/system suppliers
- Inspection of facility and equipment systems
- Operator training
- Functional testing
- Assembly of documentation
- As-built drawings
Many of the tasks involved in the commissioning and start-up of a project are also needed as part of the validation program. Overall planning will prevent double handling of information or the possibility that information provided by a contractor could get lost in the transition.
The schedule for commissioning and start-up should be integrated with the schedule for validation. At the end of this phase, the facility and manufacturing processes are turned over to the owner by the contractor.
Preparing and Executing Validation
Portions of the process, manufacturing equipment, and facility will be specified, designed, and constructed to comply with the FDA and/or Pharmaceutical Inspection Co-operation Scheme (PIC/S) current Good Manufacturing Practices (cGMPs) as appropriate. As part of cGMP compliance, qualification and validation studies will need to be performed. The purpose of qualification/validation is to develop documented evidence that only what is from a risk-based point of view is critical: process equipment, utilities, and support services processes. All these can then be performed within a CQV approach and be reliably, repeatedly, and reproducibly verified by CPV program.
The qualification of process and facility systems should be scheduled based on their completion. Qualification will overlap with commissioning and start-up of the systems. Some systems may be validated simultaneously, whereas the testing of others may be dependent on the previous completion of testing of support/utility systems that supply them. For example, media fill tests cannot begin until a sterilizer is qualified, and the sterilizer cannot be qualified until the clean steam generator supplying it is qualified.
The validation program involves the challenging (testing) of all services and equipment using appropriate methodologies and comparing the results with acceptance criteria described in previously approved qualification/validation protocols. Representatives from QA/QC, production, material science and technology (MS&T), and engineering are involved in the review and approval of all qualification/validation protocols prior to their execution. These same representatives will also review all final documentation and reports on completion of the test work.
A complete qualification package will include an installation qualification (IQ) and an operational qualification (OQ) and may include performance qualification (PQ) protocols. An IQ and OQ will be performed for all services and equipment, whereas a PQ will be performed only for those systems or processes that may affect the characteristics, quality, efficacy, and safety of the final drug product(s).
Installation Qualification
During the IQ, a complete review of the installed system (service or equipment) will be performed. The protocol will provide a systematic method to check the system’s static attributes prior to normal operation. A detailed discussion of the system will be written and should include a description of what the system is intended to do and all its major components. The system will be reviewed following the completion of installation to verify that the system is the same as what was specified. System engineering drawings, manuals, data sheets, and specifications (URS) will be used to document proper component installation and placement. The location of the system drawings, manuals, data sheets, and specifications will be documented. The system will be examined for proper connections and installation of the supporting services (e.g., water, steam, and electric) and components (e.g., filters, coils, fans, piping, gauges, valves, and controls).
Any exceptional conditions noticed during the IQ will be identified for review on a deviations list. Exceptional conditions will be investigated and the appropriate course of action (explanation, correction, requalification studies) determined. All test data will be documented. The data collected during the IQ will be packaged and summarized either individually or as part of an IQ/OQ/PQ package for presentation, review, and approval.
Operational Qualification
On satisfactory completion of the IQ, the OQ will be performed. The OQ will describe the operational tests to be undertaken, important measurements to record, and control tolerances of parameters critical for the proper operation of the system. Test objectives, methodologies, and acceptance criteria will be defined. Calibration of the critical instrumentation in the system will be documented during the OQ. Execution of the OQ will involve testing and measuring. Controls will be manipulated to test the ability of the system to provide proper temperature, humidity, flow, etc. Indicators, recorders, and interlocks will be monitored. “Worst case” or “peak load” challenges may be defined and incorporated into the testing strategy to challenge the system's capacity. Any exceptional conditions found during the OQ will be identified for review on a deviations list and the same steps followed as those for investigating and determining about exceptional conditions, etc.
Performance Qualification
After completion of the IQ and OQ, the PQ will be performed for those systems requiring it. The PQ will be used to test systems whose operation would affect the product(s). The PQ will integrate procedures, personnel training, materials, equipment, and processes. Test objectives, methodologies, and acceptance criteria will be defined prior to execution. A sufficient number of replicate studies will be performed to determine the ability of the system or process to achieve reproducible results. Testing may include analysis for chemical, physical, and microbiological constituents and will challenge the ability of the system or process to perform the intended function. The protocols may incorporate “worst case” or “peak load” challenges into the intended operating range of the system or process. Exceptional conditions and the other steps described above will be followed.
Process Validation
After the utilities and equipment have been qualified, the validation protocols and programs for the products prepared in the facility are performed. These studies will include facility cleaning and sanitization, process equipment changeover cleaning, filter integrity, process simulation media fills, and the actual process validation studies. By their nature, these studies integrate the facility, equipment, processes, and trained operators and are conducted by the users of the facility.
Conclusion
Using the phased approach for projects discussed in this article will help to produce the highest quality project while meeting stated objectives. We have observed reductions of 30% or more compared with the usual project time frames by using the streamlined phased and risk-based prioritized approach described here. The key to successful project execution is to identify and understand the relationship between different aspects of the project (Figure 1). The integration and streamlining of activities in project execution represent a solid plan for a successful outcome and exceptional drug product(s) delivery to the patient.
Project selection takes on different faces in different corporations. While the primary goal of any project is to ensure product availability to patients at sustainable and profitable levels, some projects will focus on industrial processes and others will focus on commercial processes. From the authors’ point of view, projects must be linked to the highest levels of strategy in the organization and must be in direct support of specific business objectives.
In this difficult time, project execution is more important than ever. Projects can range from building or appropriating manufacturing sites and hospitals in just a few days to allocating limited quantities of lifesaving equipment. The biopharma industry has played a critical role in responding to the crisis. The use of phased- and risk-based approaches is able to provide the global supply of state-of-the-art new therapeutics.
The original article from 1997 is available here: ispe.org/pharmaceutical-engineering/november-december-2021/1997-article-phased-approach.
About the Authors





