Final Preparation of ATMPs at Point of Care

Advanced therapy medicinal products (ATMPs), which include cell and gene therapy (C>) products, frequently require handling steps between quality control release and patient administration. These steps take place directly at the point of care and are especially critical for C>s with limited shelf life after preparation.
Regulators expect these activities to be performed within a Good Manufacturing Practices (GMP)-compliant environment, which introduces significant practical challenges for ATMP development and creates access barriers for patients. Why? Suitable GMP facilities are often unavailable near the point of care (POC).
Regulatory Flexibility
This article calls for regulatory flexibility that permits the final preparation of ATMPs at the POC, or outside of GMP. This should be done using a framework of quality assurance measures that uphold patient safety and product consistency. Key quality measures would include:
- Only nonsubstantial manipulations, i.e., not considered GMP manufacturing steps, should take place at the POC for preparation of the ATMP.
- Generation of data should demonstrate robustness and consistency of the preparation process.
- Standardized, regulatory-approved instructions should be used at POC, adhering to good clinical practices (GCP).
- Training, accreditation, and oversight of personnel involved in the preparation process should be comprehensive.
Global Harmonization
Global harmonization of quality standards for the final preparation of ATMPs prior to patient administration is vital for fostering faster, more widespread development and access to these therapies across regions. Consistent and transparent regulatory requirements for ATMP handling at the POC can help streamline the development pipeline and reduce barriers to patient access. Clear guidelines would mitigate the regulatory uncertainty surrounding POC preparation, which can otherwise delay product availability and limit market expansion. Harmonized standards would help accelerate ATMP accessibility globally, improve manufacturing predictability, and enhance patient safety by ensuring consistent quality and handling protocols across various healthcare settings.
Background
ATMPs offer transformative potential for treating or even curing diseases that significantly impact patients’ quality of life. These therapies include cell-based treatments—either autologous (derived from the patient) or allogeneic (from a donor)—as well as gene therapies, which may be administered in vivo (directly into the patient) or ex vivo (cells are genetically modified outside the body before administration). For ex vivo therapies, the path from cell collection (e.g., through apheresis) to patient treatment is intricate and often time sensitive, with the reconstituted cell product typically possessing a limited shelf life. Many of these therapies also require essential handling steps post-GMP release, such as thawing, washing, or buffer exchange, at the POC immediately before patient administration.
Potential regulatory expectations of strict GMP requirements on these final processing steps create significant operational hurdles for ATMP developers and can restrict patient access to life-saving therapies, as it is often challenging to locate GMP-compliant facilities near clinical sites. Therefore, it is essential to find a more adaptable regulatory approach that allows flexibility in GMP requirements for these POC processes. This flexibility would enable broader access to ATMPs, ensuring timely patient access to these advanced therapies while maintaining product quality and safety through tailored quality assurance measures.
A 2023 survey conducted by the Biotechnology Innovation Organization (BIO) highlighted that the majority of processing steps for ATMPs performed at the POC are carried out in either open or hybrid systems by trained hospital personnel. These personnel follow established, regulatory-approved instructions that adhere to GCP. This ensures a consistent and standardized approach to handling. Survey respondents noted that for open systems, additional testing is commonly required to verify product quality and safety, reflecting the higher risk of contamination in an open environment. In contrast, testing requirements for closed systems were reported to be less frequent due to the reduced exposure to environmental factors, which inherently decreases the risk of contamination.
This distinction in testing requirements between open and closed systems underscores the need for a risk-based regulatory framework that accounts for system type and associated contamination risks. Such a framework would provide flexibility and support safe and efficient ATMP preparation at POC, ultimately facilitating broader patient access to these therapies while maintaining rigorous quality standards.
Regulatory Frameworks
Current regulatory frameworks often lack clear guidance on the handling steps that are required after quality control (QC) but before the administration of ATMPs to patients, leaving sponsors uncertain about specific compliance requirements. Terminology for these critical processing steps varies widely across regions, with terms like “minimal manipulation,” “preparation and modification steps,” “processing steps,” and “reconstitution activities” used interchangeably. This articles uses “processing steps.” This inconsistency complicates regulatory compliance for sponsors by creating ambiguity around the necessary compliance pathways. The following is an outline of the major regulatory frameworks that currently serve as references for maintaining compliance in ATMP processing at the POC following batch release.
US Food and Drug Administration (FDA)
In January 2020, the US FDA released guidance titled “Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs),”1 outlining requirements for gene therapy products that undergo additional handling after thawing. Additional release tests, including sterility and identity assessments, may be necessary to ensure the product’s quality and safety if post-thaw manipulations, such as washing or culturing, are performed. This is especially true within an open system. The guidance also indicates that rapid sterility testing may be permitted for products with a short shelf life, particularly for ex vivo genetically modified cells that are either administered fresh or have a limited timeframe between final formulation and patient administration.
In recent discussions, the US FDA highlighted that certain activities carried out at the POC might be classified as manufacturing. These discussions include the FDA Center for Biologics Evaluation and Research(CBER) Office of Tissues and Advanced Therapies (OTAT) Town Hall on Cell Therapy Chemistry, Manufacturing, and Controls held 7 December 2022;2 the follow-up Town Hall on 8 June 2023;3 and the BIO/FDA Liaison meeting on 8 September 2023.
This classification applies particularly to procedures involving significant manipulation or those performed in open systems, which could trigger the need for additional on-site testing such as sterility, endotoxin, and identity assessments. To enhance compliance and protect product integrity during clinical investigations, the US FDA has advised sponsors to minimize handling steps at clinical sites following the manufacturing process. This approach is intended to streamline operations and ensure the highest quality standards are upheld throughout the therapy’s life cycle.
In September 2020, the US FDA released an updated guidance titled “Regulatory Considerations for Human Cells, Tissues, and Cellular and Tissue-Based Products: Minimal Manipulation and Homologous Use.”4 This revision supersedes the previous version issued in November 2017, which was later amended in December of the same year. The primary objective of the new guidance is to clarify the definition of minimal manipulation as outlined in CFR – Code of Federal Regulations Title 21, Section 1271.3.5 This clarification aims to assist stakeholders in effectively navigating the regulatory landscape surrounding human cells and tissues.
According to 21 CFR, Section 1271.3,5 minimal manipulation is defined as follows:
- For structural tissue, any processing that does not change the original relevant characteristics of the tissue related to its function in reconstruction, repair, or replacement
- For cells or nonstructural tissues, any processing that does not alter the relevant biological characteristics of those cells or tissues.
The 2020 guidance4 highlights that the original relevant characteristics of structural tissues are defined by the properties present in the donor that contribute to the tissue’s functionality. Similarly, the biological characteristics of cells or nonstructural tissues include donor properties that influence their functions. Any alteration of these original characteristics raises safety and effectiveness concerns, making it difficult to predict how the product will perform after transplantation.
Consequently, determining whether human cells, tissues, and cellular- and tissue-based products (HCT/P) are minimally manipulated relies on the impact of manufacturing on these original characteristics in the donor, rather than on the intended use in the recipient. It is crucial to note that if there is insufficient information demonstrating that the processing aligns with the definition of minimal manipulation, the US FDA will classify the processing of a HCT/P as “more than minimal manipulation,” thereby subjecting it to more stringent regulatory requirements.
A session entitled “Quality Standards for Final Preparation of Cell and Gene Therapy (C>) Products at POC” took place at the BIO International Convention in San Diego, California, in June 2024.6 During this session, the US FDA discussed their vision for the future of C> manufacturing, highlighting that much of the production may take place at clinical sites. The following outlines the main highlights of the feedback received by the US FDA:
- The US FDA provided the example of using aseptically maintained closed systems for the manufacturing of CAR T cells directly at the clinic.
- For post-release manipulations at POC, the US FDA emphasized that risk assessment concepts remain relevant and should be applied based on the specific activities performed, the environment in which they occur, and the existing controls, including personnel training, accreditation, and oversight.
- For post-release testing following thawing, the US FDA indicated that sponsors must demonstrate effective control over their processes.
- The US FDA acknowledged that current standards need to be refined to better accommodate the unique challenges posed by advanced therapy products. For instance, waiting 14 days for sterility test results for products with a limited shelf life is impractical.
The US FDA also urged for greater collaboration between industry stakeholders and regulatory bodies to develop effective solutions that address these issues and enhance the overall framework for managing advanced therapies at the POC.
United States Pharmacopoeia (USP)
A similar approach is outlined in United States Pharmacopeia (USP) <1046> “Cell-Based Advanced Therapies and Tissue-Based Products,” which underscores the essential elements of clinical site preparation and administration. The guidance states that “before the administration of certain cell or tissue-based products, one or more product modifications or preparative steps may be required.”7 These modifications often occur close to the time of administration, potentially placing them outside the direct control of the original manufacturer.
USP <1046>7 further elaborates on the nature of these product modifications or preparative steps, which may include:
- Change in final container: The manufactured product may need to be transferred from its original storage or transport container to a different one for administration.
- Change in physical state or temperature: Certain products may require thawing or warming to achieve the appropriate state for use.
- Change in solution or suspension: Some products may need to be dissolved, diluted, or suspended in a suitable liquid prior to administration.
- Combination with a biomaterial: Therapeutic cells might be combined with scaffold materials such as decellularized extracellular matrix sheets, gels, or other forms of biomaterials. Additionally, cells may be added to preexisting medical devices, like hollow-fiber filtration units, before use.
- Admixture or compounding: In some instances, mixing or compounding of cell products at the clinical site may be necessary.
- Filtration or washing: If the manufactured product contains unwanted materials, such as particulates or cellular debris, washing or filtration steps may be required to ensure purity.
- Sampling: Prior to administration, sampling of the final product may be needed to test the final formulation.
Furthermore, it is advised to monitor cell viability and potency post thawing for informational purposes, but release testing is not required at this stage before clinical deployment. Nevertheless, USP <1046> highlights that “cell-based therapy products that are prepared or modified at clinical sites need to be checked or tested appropriately to ensure they meet all quality specifications before being released for patient treatment.”7 The extent and nature of these manipulations will determine whether additional release requirements or critical specifications are necessary beyond those established immediately after initial manufacturing. Thus, the need for further release testing of “clinical site–manipulated cell products” should be assessed based on the specific nature and extent of the manipulations performed.
European Commission and European Medicines Agency (EMA)
The EMA guidance titled “Guideline on Quality, Non-Clinical and Clinical Requirements for Investigational Advanced Therapy Medicinal Products In Clinical Trials,” 8 along with the European Commission’s EudraLex document, “The Rules Governing Medicinal Products in the European Union, Volume 4: Good Manufacturing Practice, Guidelines on Good Manufacturing Practice Specific to Advanced Therapy Medicinal Products,”9 delineates essential considerations regarding processing steps in the context of ATMPs.
These guidelines specify that processing steps involve actions taken after batch release and before administration to patients, which may take place outside of a GMP environment. A key point made in these guidelines is that any procedure involving substantial manipulation cannot be classified as processing steps. Each step in processing must be accompanied by a justification for why those activities were not completed during the manufacturing phase prior to batch release.
The European Commission’s EudraLex document9 lists the following examples of processing steps:
- Thawing and washing: This involves thawing the product, washing it, and performing buffer exchange, including centrifugation to eliminate preservation solutions—such as dimethyl sulfoxide (DMSO)—and to remove process-related impurities, such as residual preservation solution and dead cells, potentially through filtering.
- Resuspension and dilution: This step entails resuspending, dissolving, or diluting the product with an appropriate solvent or buffer to prepare it for administration.
- Mixing: The product may need to be mixed with the patient’s own cells, an adjuvant, or other substances required for administration, including matrices. However, it is crucial to note that mixing a gene therapy vector with autologous cells is classified as a manufacturing activity and must be conducted under GMP.
- Splitting doses: This includes dividing the product for separate doses and adjusting dosages based on specific requirements, such as cell count.
- Loading for delivery: This encompasses loading the final product into delivery systems or surgical devices and transferring it to an infusion bag or syringe for administration.
For authorized ATMPs, it is essential that the processing steps are validated to ensure that these steps do not adversely affect the quality, safety, or efficacy profile of the ATMP. Notably, such processing step activities are not deemed substantial manipulation, and the associated risks are generally considered lower compared to GMP manufacturing processes involving more complex substantial manipulation.9
Pharmaceutical Inspection Co-Operation Scheme (PIC/S)
According to the PIC/S “Guide to Good Manufacturing Practices for Medicinal Products. Annex 13.”10 processing steps of investigational medicinal products (IMPs) is defined as a process that is generally not regarded as manufacturing, unless specific national laws state otherwise. As such, this guideline does not encompass processing steps activities. The term “reconstitution” refers specifically to the straightforward process of dissolving or dispersing an IMP for administration to a trial subject. This may also include diluting or mixing the investigational product with other substances that act as a vehicle for its delivery.
It is essential to clarify that reconstitution or processing steps do not involve the mixing of various ingredients—including the active substance—to create the IMP itself. Instead, for a process to be classified as a processing step, the investigational medicinal product must already exist prior to the procedure. Additionally, the processing steps should ideally take place as close to the time of administration as possible to ensure product integrity and efficacy. This procedure must be thoroughly outlined in the clinical trial application dossier and made readily accessible at the clinical trial site, ensuring that all personnel involved are aware of and adhere to the defined protocol.
This distinction between processing steps and manufacturing is critical for compliance with regulatory expectations and maintaining the quality and safety of investigational therapies. By clearly defining the parameters of processing steps, the PIC/S guidelines aim to provide a framework that supports the safe and effective administration of IMPs while minimizing the risks associated with handling complex therapeutic products. Proper documentation and adherence to established protocols can help ensure that processing steps do not compromise the integrity of the investigational medicinal product, ultimately contributing to the success of clinical trials.
World Health Organization (WHO)
In May 2023, the WHO released a technical document titled “Considerations in Developing a Regulatory Framework for Human Cells and Tissues and for Advanced Therapy Medicinal Products, Annex 3.”11 This document aims to support regulatory convergence and reliance for human cell therapies and ATMPs worldwide, promoting consistent regulatory practices across jurisdictions. By fostering a globally harmonized regulatory framework, the WHO seeks to facilitate the development, safety, and accessibility of these advanced therapies. The guidance provides a comprehensive foundation for the effective oversight of HCTs and ATMPs, including definitions, product categorization, and a risk-based approach to regulation, each designed to ensure quality, safety, and efficacy.
The WHO document specifies that minimal manipulation involves processing cells or tissues in ways that do not significantly modify their original structural integrity, functional properties, or safety profile. This definition is intended to differentiate between simple, low-risk processing and substantial manipulation, which requires greater regulatory scrutiny. Minimally manipulated cells and tissues are expected to perform their primary function locally without a systemic effect and must rely solely on their inherent metabolic activity.
Recognized minimal manipulation procedures include sizing, rinsing, and saline washing. Additionally, certain regional regulatory frameworks may permit more extensive procedures under the minimal manipulation classification, such as cutting, grinding, centrifugation, freeze-drying, antibiotic treatment, sterilization, and cryopreservation, depending on the risks associated with these processes and their impact on the tissue’s structural and functional characteristics.
In the context of POC applications, minimal manipulation is crucial for defining permissible on-site processing steps that healthcare providers can perform immediately before administration without needing a full GMP environment. By limiting POC activities to minimal manipulation, the WHO guidance could be applied to reduce the complexity and cost associated with manufacturing ATMPs while ensuring that the quality, safety, and efficacy of the product remain intact for the patient’s benefits.
Risk Mitigation Approaches
A comprehensive risk assessment that encompasses all processing steps, post-release testing, and pre-administration activities is essential. This helps ensure that these procedures do not adversely impact the quality, safety, or efficacy of C> products when conducted at the POC. Each preparation step should undergo an evaluation to identify potential risks and relevant mitigations needed to maintain product integrity and patient safety. The importance of this risk assessment is underscored in various regulatory guidelines, including PIC/S Annex 13,10 ICH Q9(R1),12 and associated GMP standards. In addition to the risk assessment, site qualification is a critical factor for the success and regulatory compliance of POC administration. Key components of site qualification include the following.
Personnel Training
It’s necessary to ensure that all personnel involved in product handling and administration are adequately trained in both site-specific and sponsor-specific standard operating procedures (SOPs). This training should cover all relevant protocols for preparation, storage, handling, and administration of the product, with a focus on maintaining quality and compliance with relevant SOPs.
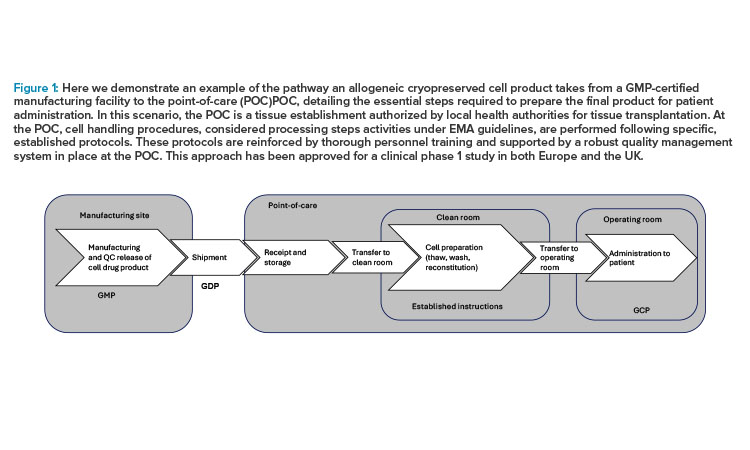
Figure 1: Here we demonstrate an example of the pathway an allogeneic cryopreserved cell product takes from a GMP-certified manufacturing facility to the point-of-care (POC)POC, detailing the essential steps required to prepare the final product for patient administration. In this scenario, the POC is a tissue establishment authorized by local health authorities for tissue transplantation. At the POC, cell handling procedures, considered processing steps activities under EMA guidelines, are performed following specific, established protocols. These protocols are reinforced by thorough personnel training and supported by a robust quality management system in place at the POC. This approach has been approved for a clinical phase 1 study in both Europe and the UK. Novo Nordisk ®
Qualification of Process Steps
Qualification of steps performed at the POC is critical in ensuring the safety, efficacy, and consistency of ATMPs. Because POC environments are less controlled than centralized manufacturing facilities, validating each step of the preparation process helps confirm that products meet the required quality standards. Furthermore, procedures implemented at the POC site should include detailed instructions for the storage and handling of the product to prevent degradation or contamination. This should include protocols to maintain complete traceability, especially for autologous products.
For autologous cell therapies, traceability is particularly crucial to ensure that products are accurately matched to each individual patient. It prevents cross-contamination or misidentification. This comprehensive approach to risk assessment, site qualification, and procedural compliance helps safeguard the therapeutic efficacy and patient safety of ATMPs administered at the POC.
Case Study: Allogeneic Cryopreserved Cell Therapy Medicinal Product
Figure 1 provides an example of the pathway an allogeneic cryopreserved cell product takes from a GMP-certified manufacturing facility to the POC, detailing the essential steps required to prepare the final product for patient administration. In this scenario, the POC is a tissue establishment authorized by local health authorities for tissue transplantation. At the POC, cell handling procedures, considered processing steps activities under EMA guidelines, are performed following specific, established protocols. These protocols are reinforced by thorough personnel training and supported by a robust quality management system in place at the POC. This approach has been approved for a clinical phase 1 study in both Europe and the UK.
Rationale for Processing Steps at POC
The rationale for conducting processing steps at the POC relies on maintaining the high quality and stability of the cell-based drug product during transport and storage, and ensuring it is safe for patient administration. Cryopreservation is essential to protect cells through transit to the POC and during storage prior to use. However, after thawing at the POC, the drug product cannot be administered directly from the vial because the cryopreservation medium contains DMSO and other cell culture reagents, which are not suitable for direct injection into patients. If these components are not removed and replaced with an excipient-grade buffer appropriate for human administration, they could lead to local toxicity at the injection site, posing significant safety risks to the patient.
Thus, washing and reconstituting the cells at the POC is a critical step. This process removes the cryopreservation medium, minimizing potential adverse reactions and ensuring the drug product meets necessary safety standards. Processing steps at the POC also helps maintain cell viability and therapeutic functionality, as the cells can be quickly prepared in a controlled environment and administered shortly thereafter, reducing the time between thawing and patient use. By performing these steps at the POC, healthcare providers can more effectively manage risks associated with cell handling and enhance the overall safety and quality of the final product administered to patients.
Cell Handling Strategy
The processing steps for the drug product at the POC involve a streamlined process of thawing, washing, and resuspension, specifically designed to minimize handling and reduce the potential for error. The washing step is implemented immediately after thawing to effectively remove the cryopreservation medium, thereby preserving cell integrity and quality. Following washing, the resuspension step adjusts the cells to the intended concentration, ensuring accurate dosing for patient administration. This handling procedure is classified as simple aseptic processing and is performed under the quality system already established at the POC tissue facility. The controlled environment, combined with trained personnel and procedural oversight, ensures that these processing steps are carried out with high precision and safety.
| Step | Risk | Mitigations and Control | Responsibility |
|---|---|---|---|
| Receipt | Traceability | Unique numbering of each drug product vial | POC |
| Information technology (IT) system (RSTM) to manage medication dispensing per study participant | POC | ||
| Established procedures for bidirectional tracing back to donor | POC | ||
| Label control according to trial medication manual | POC | ||
| Use of established cell handling instructions, including documentation of each step in formulars and QC review of these before patient administration | Sponsor/POC | ||
| Storage | Stability (i.e., the preestablished drug product stability may be compromised) | The drug product vials are transported using a designated and validated precooled transporter to the nitrogen tank for storage until use | POC |
| Visual inspection that the vials are still frozen when removed from transporter | POC | ||
| Thaw | Potency/strength (i.e., the cell concentration and viability may be incorrect) | Thawing performed in an automated thawing instrument | POC |
| Cell concentration and viability assessed prior to patient administration | POC | ||
| Washing and resuspension | Microbial safety (i.e., the drug product may be contaminated during aseptic cell handling) | Qualification/training of personnel in the aseptic cell preparation process | Sponsor/POC |
| Establishment and use of cell handling instructions, including documentation of each step in formulars and QC review of these before patient administration | Sponsor/POC | ||
| Handling of the cells in a controlled environment | POC | ||
| Rapid microbial test and compendial sterility test of the spend washing solution | Sponsor/POC | ||
| Follow-up procedure in case of positive microbiological test result | Sponsor/POC | ||
| Prophylactic antibiotic treatment and careful clinical monitoring of the study participants | Sponsor/POC | ||
| Potency/ strength | Reduced cell count, viability, homogeneity in suspension, and stability | Qualification of personnel | Sponsor/POC |
| Establishment and use of cell handling instructions, including documentation of each step in formulars and QC review of these before patient administration | Sponsor/POC | ||
| Equipment in control | POC | ||
| Verification of cell count and viability post cell handling | POC | ||
| In-use stability studies covering cell preparation procedure | Sponsor | ||
| Resuspension of cell suspension just prior to loading the device | POC | ||
| A final QC review of cell preparation documentation before administration | POC |
Risk Evaluation
A thorough risk assessment was conducted using the failure mode and effects analysis (FMEA) method, which involved evaluating and assigning ratings for the severity and probability of occurrence of each identified risk, following the guidelines set forth in ICH Q9 R1 and ISO 14971.12, 13 This assessment highlighted several key risks, including:
- Microbial safety: The potential for microbial contamination during the aseptic handling of cells poses significant risks to the safety of the product.
- Potency and strength: Risks associated with inaccurate cell counting, diminished cell concentration or viability, inconsistency in cell suspension homogeneity, and reduced stability of the drug product can severely impact its effectiveness.
- Traceability: Ensuring the traceability of vials from the moment they arrive at the POC until administration to the study participant is crucial for maintaining oversight and accountability.
To effectively address these risks, various mitigation strategies were put in place, focusing on ensuring thorough personnel and site qualification. This involved the development of specific process instructions and documentation templates, with confirmatory QC testing of the cell product prior to its use. Table 1 provides a detailed overview of the risks associated with each stage of the process, the corresponding mitigation strategies, and the designated roles responsible for implementing these actions.
Site Qualification
The POC site serves as a licensed tissue establishment authorized by local health authorities to manage and perform tissue and cell transplants. A thorough qualification process was undertaken to ensure that the site meets stringent quality standards. This involved an auditing procedure by the sponsor, which confirmed that the POC had implemented a robust quality management system. The findings indicated that the facility is equipped with a controlled environment tailored for aseptic cell handling, supported by the necessary SOPs designed to maintain both the facility and its equipment in a state of continuous control. This thorough assessment validated the POC’s capability to perform its intended functions safely and effectively.
Cell preparation at the POC is conducted within a cleanroom that adheres to aseptic techniques to mitigate contamination risks. The actual handling of the cell product is executed on a monitored laminar flow (LAF) bench, which provides a sterile environment essential for maintaining cell integrity. To ensure compliance with environmental safety standards, environmental monitoring is regularly performed using settle plates during process qualification runs.
These assessments have confirmed the effectiveness of the LAF bench in providing a sterile environment. They support the overall safety and quality of the cell preparation process prior to patient administration. This meticulous approach underscores the commitment to maintaining high standards in the handling and processing of cells at the POC.
As ATMPs continue to advance in medical innovation, the capability of community hospitals to administer these treatments becomes crucial.
Qualification of Cell Handling and Staff Training
Several qualification runs were performed by two separate operators, and the sterility of the handled cell product was validated using the European Pharmacopoeia (Ph. Eur.) compendial sterility method. To further ensure the integrity of the aseptic handling procedures, the team conducted aseptic process simulations, commonly referred to as media fills. Personnel tasked with cell preparation underwent extensive training focused on aseptic techniques, washing, and resuspension processes. They were also trained on the specific cell preparation protocols and had to demonstrate their proficiency in producing a sterile cell product that met the required concentration and viability standards.
Assuring Traceability
At the POC, a comprehensive bi-directional traceability system was established to track each patient’s treatment back to the original donor. Each vial is equipped with a unique identification number, and an integrated IT system was implemented to oversee the dispensing of medication for all participants. This system offers the sponsor detailed visibility throughout the entire life cycle of the vials, encompassing storage, transit to the POC, receipt, dispensing for clinical administration, and thorough drug accountability and vial reconciliation. Additionally, label control processes adhere strictly to the guidelines outlined in the trial medication manual and the cell handling instructions, with every manual entry subject to verification by a second individual to guarantee precision and regulatory compliance.
Processing Steps at the POC
The following instructions specific to the cell handling procedure were included in the clinical trial application to ensure a standardized approach during the preparation of the drug product for study participants.
Preparation of resuspended cell product for administration
This instruction aims to guide personnel responsible for the thawing, washing, and resuspension of the drug product prior to transplantation. It outlines the necessary steps to ensure successful preparation and requires documentation of adherence to the procedure. The instructions are printed and utilized as a participant-specific preparation record during the handling of the trial product. Upon completion, the document and its attachments must be filed in the investigator trial master file. In cases where deviations occur during cell preparation, the individual in charge of cell handling must consult with the designated approver before proceeding further.
Instructions for handling resuspended cell product in the operating room
This section details the necessary steps for loading the cell suspension into the injection device within the operating room. The medical doctor responsible for the transplantation procedure oversees this process. Additionally, the cell handling personnel designated at the study site must possess prior experience with cell handling and have undergone training specific to the procedures outlined in the study protocol. This ensures that all actions taken during the cell handling process are executed with the utmost care and expertise.
Testing of the Final Product
To ensure cell quality with regard to sterility, both a rapid microbial test and a compendial sterility test were performed on spent media collected during the cell washing step. The rapid microbial test provided an immediate indication of any microbial contamination, preventing the administration of an unsterile product. Given that the compendial sterility test results would only be available after administration, any positive sterility test result would be logged as a serious adverse event.
In such cases, the participant would immediately begin appropriate antimicrobial treatment, and the treatment of new patients would be paused until the contamination source was identified and corrective actions were implemented. Verification of cell concentration and viability was conducted on the final cell suspension after all cell handling steps but before patient administration. If either cell concentration or viability fell outside the acceptable range, the cell suspension would be discarded to maintain product safety and efficacy.
Challenges for Processing Steps at the POC
Implementing GMP standards for processing steps at the POC involves a careful balance of patient safety and accessibility challenges. Although GMP compliance aims to reinforce patient safety and product quality, it may inadvertently limit patient access to advanced therapies. First, only a limited number of hospitals are equipped to meet stringent GMP standards, reducing the availability of cell therapies to fewer facilities. This limitation is especially impactful in rural or underserved areas, where access to GMP-compliant facilities is scarce. Additionally, patients may face longer travel distances to reach GMP-certified centers, creating obstacles for those with mobility constraints or limited financial resources.
Adding to this complexity, regulatory expectations for C> processing vary widely across global jurisdictions, driven by differences in healthcare systems, regulatory frameworks, and risk management priorities. For instance, the EMA allows some POC processing activities to be performed outside of GMP, a decision that supports faster access to these critical therapies. This approach contrasts with other regions where GMP requirements are more stringently enforced, regardless of the product’s location or processing steps needs.
The different characteristics of each cell therapy product and related POC handling strategy pose a challenge to harmonization of POC regulatory expectations. Nevertheless, such harmonization is indeed needed to minimize the current variations across POC sites due to different practices and expectations in different regions, and it would help assure that the reconstitution activities do not impact safety and efficacy of the product administered to patients across countries.
Quality and Safety
According to a 2023 survey conducted among BIO members, processing steps at the POC site are primarily performed outside traditional GMP facilities, often in open or hybrid systems. To uphold the quality and safety of the final product prior to patient administration, it is essential to establish robust procedural, handling, and environmental controls. Key strategies to achieve these controls involve:
- Applying GMP and GCP principles
- Using closed systems wherever possible
- Maintaining aseptic conditions during open processing steps
- Following strict written procedures with thorough documentation
Staff Training
Staff training is also fundamental to maintaining these standards and ensuring procedural compliance. Currently, the administration of commercially available ATMPs is limited to a select number of tier 1 FACT-accredited transplant centers and academic medical centers within the US, along with similar high-standard facilities worldwide.
This limitation poses significant access barriers, as only a fraction of patients have access to these specialized centers equipped for ATMP delivery. To address product quality assurance at POC, the US FDA has indicated that additional release testing, including sterility and identity assessments, may be necessary to validate these therapies prior to administration. This further underscores the critical need for stringent quality measures outside of traditional GMP environments.
Sterility Testing Exemptions
Under 21 CFR 610.12 and 21 CFR 1271.155,,14 , 15 cell therapy products may be exempt from sterility testing if processed in closed systems without manual handling, pending a case-by-case risk assessment. However, most POC processing, especially during early clinical phases, involves open or hybrid systems, which makes meeting these exemption criteria challenging. To navigate this, detailed guidance documents are needed to support consistent application of sterility standards. Such documents should outline specific controls and testing strategies tailored for open and hybrid processing systems, helping POC facilities maintain product quality and expand access to cell therapy treatments while adhering to essential regulatory standards.
When determining sterility testing requirements, a comprehensive approach is critical. This involves integrating microbial contamination risk mitigation strategies, such as robust process controls, continuous active monitoring of aseptic practices, environmental monitoring, and the use of suitable facilities. Additionally, a rigorous training program for personnel engaged in aseptic processing is crucial, especially given that sterility testing results often become available only after patient administration. Establishing these layers of control ensures that even without immediate sterility results, the final product meets safety standards, reducing patient risk associated with microbial contamination.
The Need for Traceability
The case study in this article highlights the critical role of a robust bi-directional tracing system that tracks cell therapy products from the patient back to the donor, ensuring identity and safety without the need for additional identity testing. At the POC, each vial is uniquely labeled for each patient, allowing for comprehensive traceability throughout the product’s life cycle—from drug product release through administration.
This patient-specific labeling and tracking system grants the sponsor and clinical teams full oversight of each vial’s journey, meeting regulatory standards and enhancing patient safety. By using automated or semi-automated systems integrated with patient records, this approach minimizes potential errors and enables swift reconciliation. Such systematic control and verification steps effectively eliminate the need for extra identity testing, as each phase of handling is safeguarded by rigorous tracking protocols.
New POC Sites
Adding new POC sites during clinical development and after marketing authorization application/biologics license application (MAA/BLA) approval is an area where regulatory flexibility is essential to support patient access without compromising product safety and quality. Introducing ATMP preparation and administration at a new POC site requires a comprehensive risk assessment to evaluate any potential variability introduced by the new setting, particularly concerning the complexity of the processing steps involved.
This risk assessment should be complemented by established procedural controls, such as standardized instructions, a robust personnel training program, and rigorous facility qualification processes. Ensuring alignment with GCP principles can help maintain high standards, ideally allowing new POC sites to operate without necessitating separate regulatory approvals. This approach supports both quality and accessibility, enabling wider patient access to ATMPs across various clinical settings.
Improvements
As cell therapy products progress through mid-to-late stages of clinical development, advancements in the processing steps often emerge. Examples of this include the development of fully closed systems or optimized cryopreservation methods. Although these improvements necessitate comparability studies16, 17 and requalification of the processing steps, they also highlight the importance of regulatory flexibility to enable seamless transitions; for instance, moving from open handling in a controlled aseptic setting to closed handling in a more adaptable environment.
Building a robust network of qualified treatment centers is essential not only for delivering consistent and high-quality care, but also for advancing the development of personalized ATMPs.
A phase-appropriate, risk-based regulatory framework that encourages developers to refine and simplify the processing steps, while prioritizing product quality and patient safety, would support these advancements. Such a framework should strike a balance between rigorous standards and practical implementation, helping both patients and developers navigate this evolving field. Ultimately, a harmonized global approach to regulatory requirements for processing steps at the POC is essential to make these advanced therapies widely accessible.
Conclusion
The uncertainties of regulatory requirements for the final processing steps of ATMPs remain a considerable barrier to making these therapies globally accessible. Establishing a regulatory framework that does not define the POC reconstitution activities as GMP production steps and allows the developers to adhere to high-quality standards—such as the established accreditation and oversight at POC sites and adherence to GCP—could greatly enhance patient access without compromising safety or product quality. This approach is particularly important for patients who face logistical or financial limitations making difficult to travel to treatment centers, thus helping democratize access to advanced therapies.
As ATMPs continue to advance in medical innovation, the capability of community hospitals to administer these treatments becomes crucial. Building a robust network of qualified treatment centers is essential not only for delivering consistent and high-quality care, but also for advancing the development of personalized ATMPs. Embracing flexibility in product preparation requirements, paired with stringent quality assurance, will support a sustainable pathway for scaling ATMP treatments, benefiting a broader patient base.
Aligning with established guidelines and adopting a phased, risk-based regulatory approach will be key in shaping a supportive regulatory landscape for C>. Through collaboration among stakeholders, this balanced approach will drive broader patient access, accelerate therapeutic innovation, and propel the field forward, ensuring more individuals can experience the transformative potential of these therapies.


