Cell Culture Media Manufacturing Controls for Bio-Manufacturing

Advanced therapy medicinal products (ATMPs) and cell and gene therapies (C>s) represent a promising medical product class that employs gene therapy, cell therapy, or tissue engineering to address various diseases and injuries. One critical aspect of ATMP and C> manufacturing is using cell culture media. With thousands of ATMPs and C>s in clinical trial phases, the role of cell culture media has become even more significant to the biopharmaceutical industry.
ATMPs and C>s are innovative therapies that can potentially provide transformative treatments for numerous conditions, including genetic disorders, neurodegenerative diseases, cancer, and cardiovascular diseases. To ensure product consistency and performance of lifesaving therapies, it is necessary to study critical cell culture media process parameters and their impact on the key culture media attributes.
There were 2,093 ongoing clinical trials globally at the end of June 2022 (phase 1: 776; phase 2: 1,117; phase 3: 200) suggesting the upcoming slew of approvals and predictable demand in their commercialization. By region, North America leads with 808 active clinical trials, Asia Pacific has 640 trials, Europe has 329, and all other regions have the remainder. 1 However, the development and manufacture of ATMP and C> products pose unique challenges, including emerging regulatory frameworks and complex manufacturing processes.
Cell culture media is crucial for ATMP and C> manufacturing, ensuring optimal cell growth and function by providing essential nutrients, growth factors, and other components. The media quality and composition can directly impact the product’s characteristics, including yield, purity, and potency.
Therapies that require ex vivo cell expansion rely on cell culture media as a critical raw material. For example, in the ex vivo expansion of human mesenchymal stem cells, well-defined serum-free formulations are desired to support efficient production while maintaining cell properties. Therefore, proper selection and optimization of cell culture media are vital. By using quality-
controlled, process-optimized media formulations and customized media, consistency and reproducibility can be maintained. This ultimately benefits patients by providing safe and effective therapies. Even with the same specifications, different media formulations from different suppliers can lead to varied cellular responses, growth patterns, and experimental outcomes. The choice of a culture medium should be based on the specific requirements of the experiment and the characteristics of the cell line being used.
Regulatory Scenario
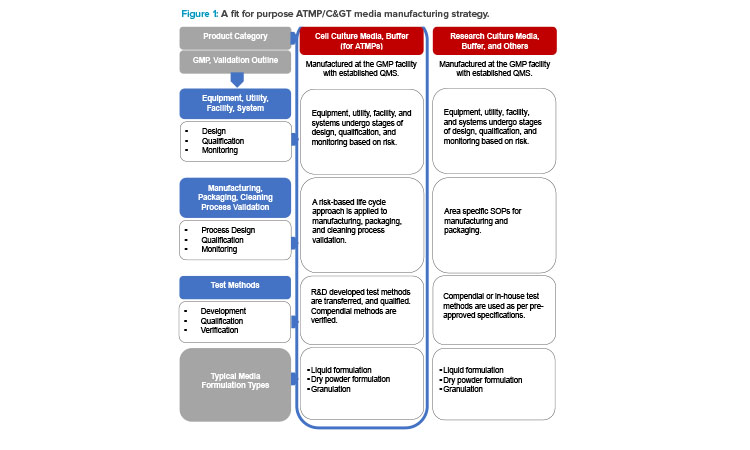
Raw and ancillary materials, like cell culture media used in manufacturing GMPs, are not explicitly discussed. However, 21 CFR Part 210 (210.3(b)3) touches on components, which these products would most align to per their definition.2 As such, there is an expectation that the media manufacturers will comply with general GMP sections. Media materials are indirectly regulated by ensuring adherence to standards outlined in the United States Pharmacopeia (USP), European Pharmacopoeia (EP), chemistry, manufacturing, and controls (CMC) guidelines, and voluntary consensus standards such as ISO.
In the US, cell culture media is classified as an ancillary material, whereas in the EU, it is considered a general raw material according to GMP guidelines. In certain cases, device guidelines, such as
21 CFR Part 820,3 may apply. Hence, standard cell culture media manufacturers apply fit-for-purpose (see Figure 1) sterile and nonsterile GMP requirements. The USP <1043> Ancillary Materials for Cell, Gene, and Tissue-Engineered Products4 compendia classifies ancillary materials (media), as shown in Table 1.4
| Ancillary Materials Risk Tier | Typical Use in Cell, Gene, or Tissue- Engineered Product Manufacturing | Suggested Qualification or Risk Reduction Activities |
|---|---|---|
2 Low-risk, well-characterized materials with intended use as | Cell culture medium additive (ex: tissue culture media) | Refer to USP <1043> |
3 Moderate-risk materials not intended for use as ancillary | Cell culture medium additives, induction agents, buffer components (ex: purified chemicals, reagent-grade) | No additional controls recommended |
Cell culture medium additive (ex: recombinant growth
| Drug master file (DMF) cross reference, certificate of analysis (CoA), lot-to-lot e ect on process performance, removal from the final product, stability assessment, functional assay, vendor audit, GMP manufacturing process, internal specs, lot-to-lot biocompatibility, cytotoxicity, adventitious agent testing | |
4 High-risk materials | Cell culture medium additive (ex: fetal bovine serum [FBS], animal-derived, including human, extracts) | Same as 3, plus verify traceability to country of origin, assure the country of origin is qualified as safe with respect to source-relevant animal diseases, including transmissible spongiform encephalopathy (TSE), adventitious agent testing for animal source-relevant viruses |
Table 1
Overcoming Risk to Final Product
Contamination
Sterility assurance is critical because it can significantly impact the growth and behavior of cells and ultimately compromise the outcomes. Contamination by microorganisms such as bacteria, fungi, and viruses can result in reduced cell viability and proliferation. Precautions such as applying aseptic behaviors and techniques, maintaining a clean environment, implementing engineering or procedural controls, and regularly monitoring for signs of contamination (i.e., contamination control strategies5) are also essential during media manufacturing.
The potential to transmit adventitious agents exists with the culture media. Hence, a risk-based control strategy must be developed. To facilitate the reduction of viral risks, it is crucial to establish strong supplier controls and perform assessments specifically aimed at clearing or reducing viral risks. Proof of viral contamination controls are reviewed as part of the regulators’ safety assessment of the finished ATMP. Therefore, it is essential to understand the impact of cell culture media components when determining the viral safety profile. Several clearance steps, such as purification and filtration, may be part of the manufacturing process. Hence, filter studies with viral spiking are part of process development.
Typically, established controls involve the analysis of viral contaminants performed at different stages under various conditions. Further, adventitious agent testing is required per 9 CFR Part 113,6 the EMA’s “Guideline on the Use of Bovine Serum in the Manufacture of Human Biological Medicinal Products,”7 and other regulations.
Animal Origin Concerns
The International Conference on Harmonisation of Technical Requirements For Registration of Pharmaceuticals For Human Use (ICH) Q5A(R1) guideline describes the viral safety testing and evaluation of biotechnology products derived from characterized cell lines of human or animal origin (i.e., mammalian, avian, and insect). It outlines the data that should be submitted. Animal origin (AO) components may impact batch-to-batch variability, depending on the source and processing method, and pose reproducibility issues, resulting in a lack of product availability. There are also associated ethical concerns and difficulties in characterization due to the existence of many proteins and metabolites.8
Once identified, these risks may be systematically addressed. However, based on the C> developer’s risk mitigation strategy and criteria, animal origin-free (AOF) media can be an option. There needs to be transparency about the basis of an AOF claim. AOF refers to the absence of materials derived from animal sources, including human sources, in a product or manufacturing process. Hence, the processing, control, and qualification measures need to be defined by the supplier. Manufacturers should have a comprehensive process to support the AO contamination controls, including ensuring that materials are not secondarily exposed to AO.
Customized Formulations for Cell Culture Media
Choosing the right cell culture media for a specific cell type can be a daunting task, but it can be made easier with the help of media formulation experts from manufacturers who can provide customized formulations when necessary. It is important to consider the compatibility of culture media components with downstream process materials and assays, such as polymerase chain reaction (PCR), enzyme-linked immunosorbent assay (ELISA), or flow cytometry, as they can interfere with the results. For this reason, adjusting various attributes of the media—such as nutrient and growth factor concentrations, trace elements, and pH—may be necessary to achieve the desired outcomes.
Maintaining the culture media at precise pH and osmolality levels is crucial for optimal cell growth and function. Manufacturers implement in-process testing controls to ensure these critical quality attributes are maintained. Buffering agents and salt concentration adjustments are used to maintain pH and osmolality ranges when needed, as fluctuations can cause cell stress and impact outcomes. Nutrient depletion can also occur in culture media, reducing cell growth and function. Stability data is generated to support proper storage, establish expiration dates, and ensure adequate storage and monitoring.
Minimizing Variability in Media Manufacturing
In manufacturing cell culture media, there can be variability in the composition and quality within and between batches, which can impact reproducibility. This variability can be caused by several factors, including the input of raw materials (material); manufacturing equipment, change parts, and product contact components (machine); process parameters (method); storage conditions (nature); test methods (measurement); and opera-tor-to-operator differences (human).
To ensure consistent and high-quality cell culture media, it is essential to understand the sources of variability and develop a robust control strategy that minimizes both within-batch and batch-to-batch variations. The total variability can be expressed as the sum of individual component variations. This may be mathematically denoted as:9
S2total =S2material +S2equipment + S2processes + S2test method +...
= S2inter-batch + S2intra-batch
Liquid Media Processing Parameter Controls
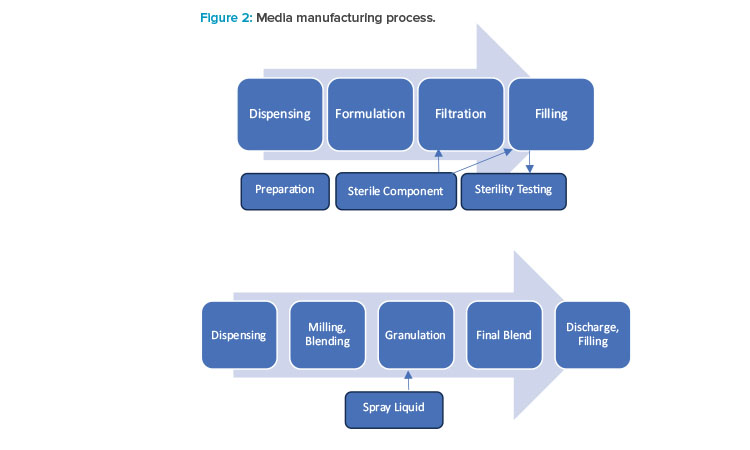
For liquid media manufacturing, a series of operations—including raw materials and water for injection dispensing, formulation, filtration, and filling—are executed with their established parameters and attributes to produce the desired quality of media product. To control the risk of cross-contamination and ensure effective cleaning, qualified inline cleaning (clean-in-place) and sanitization (steam-in-place) processes are to be employed where appropriate.
Product Sterility
During liquid filling, aseptic practices and gowning procedures are strictly followed, and cleanroom engineering controls such as classified areas and laminar airflow units are used. The product’s sterility assurance level is controlled through the operations. Product sterility can fail if sterilized components (media formulation, container, and closure) are brought together under contaminated conditions. The aseptic processing operation is qualified using a microbiological growth medium, such as soybean casein digest medium. The initial and routine media-fill program considers the risk factors for contamination on a production line and accurately assesses the state of process control following the US FDA’s “Sterile Drug Products Produced by Asep-tic Processing — Current Good Manufacturing Practice.”10
Process Parameters
The variability in the cell culture media manufacturing process is a subject for research & development and manufacturing science, and technology study during process development and technology transfer to the manufacturing floor. In particular, the process parameters for a given batch size of media formulation are examined, including weight, seal integrity, charging, temperature, tank volume, fill pump percentage, order of addition, mix time, impeller speed, dwell time, and total production volume. The impact of these parameters on the media quality attributes and the extent of the impact is then evaluated for further optimization. Design of experiments may be performed where applicable.
Subsequently, qualified recipe-based systems/skid controls are developed to deliver consistent quality liquid media for the filtration stage. The formulation tank and filtration equipment require aseptic connections and qualified filters. Where necessary, it must be purged, wetted, and tested for filter integrity using bubble point, diffusive flow, or pressure hold test. Bioburden reduction, sterility, and contamination controls are achieved through sterilization of equipment/components, the use of sterile filters, controlled environments such as a cleanroom or isolator, validated cleaning and sanitization procedures, and continual environmental monitoring and testing for microbial contamination.
Sterilization
Container closure systems—such as bottles, drums, or bags—are commonly used for liquid media and are filled using either automated/semi-automated fillers or manual filling operations. All equipment and components that contact the product during the filling process must be sterilized to minimize the risk of contamination. Aseptic techniques should be employed in applicable segments of the manufacturing operations, such as component sterility assurance, filtration, or the filling process. The filling manifolds must be wetted and purged where necessary. The auto-filler parameters may include fill volume, fill speed, time, weight, temperature, relative humidity, dosator pressure, or inert gas purging pressure.
Finishing Operations
Finishing operations are set up in a series after filling. Finishing parameters such as temperature, pressure, torque, speed, count, weight variation, induction seal power/temperature, and glue temperature are qualified. The filling assembly setup parameters for bags are also critical. To ensure product quality, a statistically sound sampling plan and sterile techniques must be used when sampling for sterility testing, routine in-process testing (e.g., appearance, conductivity, total organic carbon [TOC], pH, osmolarity), bioburden, environmental monitoring, and retention sampling. If intermediate stainless steel storage tanks or linear low-density polyethylene (LLDPE) tanks are used, hold times must be established to ensure the product is stored under appropriate conditions.
Powder and Granule Media Processing Parameter Controls
Rise in Powder and Granule Media
In the biopharmaceutical industry, the use of powder or granule media has gained popularity due to its process flexibility and elimination of excessive liquid culture media storage, transportation, and handling issues. Dry media has become the predominant type supplied on a large scale, accounting for over 90% of media used.11
The granule media format is a preferred choice among dry media formats due to its enhanced solubility profile, improved uniformity, and minimal variability achieved through granulation technologies. Granulation has allowed cell culture media manufacturers to offer a media format that can increase downstream ATMP and CG&T process efficiency, safety, and yield, surpassing the performance of traditional dry media formats. The operational consistency and reliability of the granule media format enable ATMP and CG&T developers to expedite product launches, ultimately benefiting patients.
Powder and Granule Media Process Parameters
Compared to the liquid media manufacturing process discussed previously, the powder or granule manufacturing processes have different processing parameters to be controlled, specifically for milling, blending, and granulation unit operations. The selection and design of the mill (e.g., ball mill, Fitz mill), blender
(e.g., ribbon blender, slant cone bender), and granulator are important elements and depend on the media formulation specifics. The physiochemical attributes, including particle size distribution (PSD), can vary widely, leading to challenges in media formulation at scale due to varying flow properties and powder rheology. Therefore, a comprehensive full-scale process development characterization is necessary.
Milling equipment is employed for size reduction. For example, a ball mill uses steel grinding balls to grind materials by rotating a cylinder and causing the balls to fall back onto the material. A Fitz mill uses a rotor with knives that cut and shear the material into smaller particles within a feeding chamber.
Fluid bed granulator parameters
The fluid bed granulator parameters that must be fixed or controlled include air flow, inlet air temperature, inlet air dew point, product temperature, exhaust air temperature, shake duration, shake interval, atomization air pressure, spray rate, spray quantity, and drying temperature. The inlet air temperature and velocity must be carefully controlled to ensure that the granulation process occurs at the correct rate and that the granules do not become too large or too small.
The position of the spray nozzle and the optimized spray rate can ensure that the granules are formed evenly and that the trace materials are distributed uniformly throughout the granules. The PSD of the granules is assessed. Trial runs are conducted to avoid granules being too dense or too porous. Finally, the drying conditions are optimized to achieve the desired moisture content of the granules.
Blend process parameters
Blenders, such as slant cone blenders, can ensure uniform mixing of granules. The mixing variations that occur during the final blending process can be reduced through the application of control strategies that are based on well-characterized raw material properties, robust manufacturing processes, and precise measurement systems. The granules are loaded into the blenders and are blended by tumbling as the machine rotates at a fixed speed and time on a rotational axis. The blender’s geometry is symmetric in design, with distinct hopper-shaped chambers. Sampling locations for process qualification must consider granule flow dynamics, such as those recommended in the statistically sound Grouped Area Variance Estimate (GAVE) method.12
Blend studies can establish blending process parameters, including bin fill, order of addition, revolutions per minute, and time. Operating parameters are finalized during scale-up. Controls for filling operations and discharge operations, such as discharge rate and height, are required due to the potential for variability introduction. An advantage of granulation technology is that it can produce granule media containing trace elements.
Manufacturing Process Validation
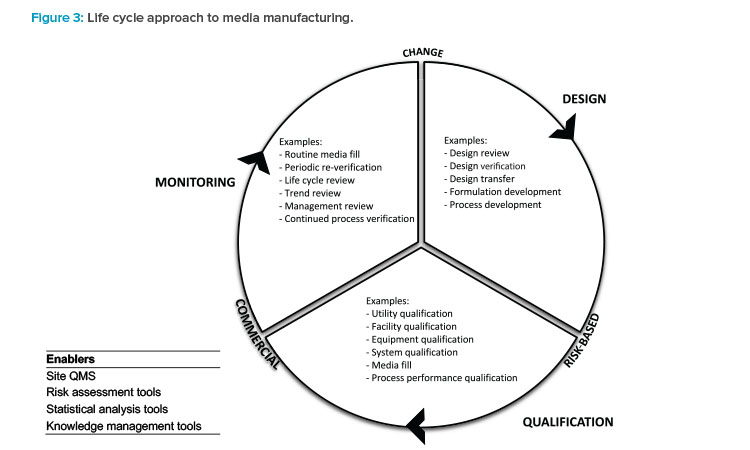
Manufacturing technology, the manufacturing process steps (see Figure 2), and formulation type significantly influence how a media manufacturing process validation approach is developed. Therefore, adopting the life cycle approach to process validation is essential, because the approach is based on sound science and data. The activities fall into three general categories of design, qualification, and monitoring (see Figure 3).
Conclusion
The development and commercialization of the cell culture media manufacturing process requires a life cycle approach.13 As such, it is important to study critical process parameters and their impact on the key culture media attributes, as well as implement a control strategy to ensure consistency and performance. The use and role of cell culture media in biopharmaceuticals are becoming significant, and the availability of media with consistent performance is essential for bringing lifesaving ATMP and CG&T products to market.
As this field continues to advance, it is crucial to remain vigilant of regulatory changes and implement robust processes that ensure the safety and efficacy of the final product. This requires collaboration between stakeholders, including regulatory bodies, researchers, and manufacturers, to establish best practices and standards that guarantee the quality and safety of cell culture media. Ultimately, these efforts will help to advance the biopharmaceutical industry further and improve healthcare outcomes for patients.
About the Authors




