Contamination Trends & Proposed Solutions
Contamination is one of the top reasons for medicinal product
To date, numerous case studies2, 3, 4, 5, 6 have been completed on contaminated medicinal products and contaminants that may be useful in identifying and evaluating methods to control and manage contamination. However, few studies have analyzed contamination trends to enable a more effective risk-based approach to control contamination in the manufacture of medicinal products.
This article aims to gather reports of contaminated medicinal products from multiple sources, such as PubMed and Embase; GMP standards adopted by the US FDA, China National Medical Products Administration (NMPA), and India Central Drug Standard Control Organisation (CDSCO); and standards from the World Health Organization (WHO) and the Pharmaceutical Inspection Convention/Cooperation Scheme (PIC/S). From the findings, the overall trends in contamination of medicinal products—including the types of medicinal products and common contaminants encountered, their causes and origins, preventive measures, and challenges faced by manufacturers and RAs—were identified and recommendations to resolve the identified problems provided.
Identification and Analysis of Contamination Trends
Three major recall databases—those of the US FDA, the United Kingdom’s Medicine and Healthcare Products Regulatory Agency (UK MHRA), and Australia’s Therapeutic Goods Administration (TGA)—were searched to assess contamination trends in the past five years. The contamination trends analysis included the year the contamination event occurred, identity of the product and contaminants/impurities, country of manufacture and product recall (if any), circumstances leading to contamination, and outcome following the contamination event. The number and breakdown by contaminants/impurities are provided in Table 1.
Unfortunately, these databases lacked information about the exact nature of the contaminant/impurity and the circumstances that led to the contamination events. To obtain deeper insight into contamination trends, PubMed, Embase, and Cochrane were searched, and cases from these literature sources were analyzed. The cases covered the same types of contaminants and impurities noted in Table 1: microbial contaminants, process-related im-purities, metal contaminants, packaging-related contaminants, drug cross-contamination, and an “unknown” category encompassing other contaminants associated with the manufacturing process, including those from cell culturing.
Microbial Contaminants
From 2007 to 2021, 90 cases of contamination due to microorganisms were reported, with 61 caused by bacteria,5, 6, 7, 8, 9, 10, 23 by viruses,11, 12 and 6 by fungi.6, 13, 14 The most commonly mentioned contaminants were Burkholderia species5, 8 as a whole, and vesivirus 2117 for Genzyme products in 2009.11
| Recalls Attributed to Contamination |
Contaminant/Impurity | ||||||
|---|---|---|---|---|---|---|---|
| Microbial | Process Related | Metal | Packaging Related | Other Drugs | Unknown | ||
| US FDA | 177 | 78 | 41 | 3 | 5 | 13 | 37 |
| UK MHRA | 67 | 27 | 27 | 2 | 2 | 2 | 7 |
| Australia TGA | 84 | 28 | 22 | - | 6 | - | 28 |
Microbial contaminants commonly occur during manufacture, often arising from the materials used. For example, bacterial and viral contaminants can occur from the use of animal sera and human plasma components.7 Bacterial contaminants are also commonly introduced into medicinal products through water-based routes, whether during manufacture of liquid preparations or from external sources.5, 8
Compounding pharmacies were commonly mentioned as sources of microbial cross-contamination, especially in the US.14, 15 The regulation of compounding pharmacies in the US has historically been murky because they are not officially considered drug manufacturers, leading to incomplete regulation and non-required adherence to GMP standards.9, 15 This has led to compounding pharmacies completing high-volume activities such as mass repackaging, thereby increasing the risk of cross-contamination. Similarly, compounding practices such as manual dilution and reconstitution10 have been associated with cross-contamination by microorganisms.
As demonstrated by the various Burkholderia cepacia outbreaks5 and the case of Streptococcus mitis/oralis-contaminated Avastin, microbial contamination has the potential to cause widespread and serious infection. The Genzyme case also demonstrates that contaminated medicinal products can lead to severe drug shortages, especially when production is monopolized by single companies.11
Process-Related Impurities
Over 30 studies were found to be related to contamination with process-related impurities.2, 4, 16, 17, 18, 19, 20, 21 More than 20 of these studies reported genotoxic impurities 2, 4, 4, 16, 17, 18, 19 such as nitrosamines2, 16, 17, 18 or ethyl methanesulfonate.4, 19 Five studies reported that the impurities were entities chemically similar to the drug substance, such as epimers, polymorphs, isomers, or drug derivatives.20
Although many of these studies did not identify the exact factors leading to contamination, the most common cause appears to be the formation of unexpected reaction byproducts during the changing of reactants during manufacture.2, 16, 17, 18 One example is the switching of tributyltin azide with sodium azide and dimethyl formamide by Zhejiang Huahai Pharmaceuticals (ZHP) in 2012 to reduce waste and to increase yield in the production of angiotensin II receptor blockers, resulting in the formation of N-nitrosodimethylamine (NDMA), a known carcinogenic impurity.18
Failure in characterizing impurities during the manufacturing stage or in the final product is another cause.19 Characterization is an important step to identify impurities and is especially crucial when manufacturers revise the manufacturing process. In ZHP’s case, omission of this step led to patients inadvertently taking NDMA-contaminated drugs for several years before the eventual detection in 2018.18
Poor cleaning practices also contribute to the formation of impurities. In the 2007 Hoffmann-La Roche Viracept incident, the holding tank was cleaned but not dried properly. This led to residual ethanol buildup and the unintentional formation of ethyl methanesulfonate.4
Although these impurities often do not pose sufficient risk to warrant a recall, mass recalls may be necessary for medicines taken for long-term use in view of the compounded risks.2, 4, 18 Patients taking these drugs may experience medication shortages, healthcare institutes may have to source safer alternatives, and RAs may be required to inspect the manufacturing premises to assess GMP compliance, suspend manufacturing, or recommend corrective actions.21
RAs may also have to review the risks of patients taking the contaminated medications,22 especially for manufacturers with large market shares (such as ZHP), which can impact large numbers of patients globally. Notably, in both the ZHP and Hoffmann-La Roche contamination incidents, despite the impurities being carcinogenic, RAs declared the risks “not significant”22 or that there were “no health risks.”4
Metal Contaminants
Of the 17 studies reporting metal contamination, various contaminants were identified. They included nickel, chromium, steel, and aluminum, as well as various metals of a wide range of weight and toxicity.23, 24, 25, 26, 27
Metallic particles that inadvertently came off the manufacturing equipment may be due to friction between two pieces of metal in the manufacturing equipment or from wear and tear during production. Noteworthy cases were the inadvertent introduction of grade 316L stainless steel into Moder-na’s COVID-19 vaccine by the outsourced manufacturer (ROVI Pharma Industrial Services S. A.25, 26 and visible black specks observed in Johnson & Johnson’s Infants’ Tylenol products.23, 24, 25 The former highlights the importance of avoiding human error in the handling of manufacturing equipment. In this case, the increased friction was caused by incorrect assembly of the manufacturing equipment due to a technician “visually misjudging the precise 1 mm gap between the star-wheel and the stopper.”25
In both recalls, metal contaminants took the form of visible “black specks” observed by consumers, which prompted further investigation into the manufacturing process. Although technology exists for the screening of elemental contaminants in pharmaceutical products,27 it appears this screening had not been done during quality control tests by manufacturers.
In the Moderna COVID-19 vaccine contamination case, three lots of the five lots affected, totaling 1.63 million vials, 26 had to be recalled and destroyed. In Johnson & Johnson’s contaminated Infants’ Tylenol incident, the manufacturer was fined US $25 million, and had to recall up to 136 million bottles of pediatric medications.23
Packaging-Related Contaminants
Eighteen studies reported contaminants from drug packaging. The contaminants included rubber, glass, plastics such as low-density polyethylene, plasticizers such as phthalates, and polymer additives such as Irganox 1010 and bisphenol A (BPA).28, 29, 30, 31
One key cause was attributed to the incompatibility between the packaging materials and the product.28 For biopharmaceuticals packed in glass vials, the strong pH and/or buffers may result in the delamination of glass, resulting in glass flakes.28 Another cause identified was poor storage conditions by manufacturers. Prolonged storage or storage at high temperatures may potentially result in container degradation and the leaching of these impurities into the product.30 For both causes, manufacturers should assess the toxicology and safety of their products in relation to the packaging materials used, as well as their storage conditions.
Many biopharmaceuticals, ophthalmics, and injectable products29, 30 are often stored in rubber-stoppered glass vials, disposable plastic syringes, polyvinyl chloride (PVC) bags, or plastic bottles. The accidental injection of rubber particles and glass flakes may lead to circulatory disorders such as vascular occlusion, ischemia, and tissue necrosis, thereby increasing the risk of embolic, thrombotic, and vascular disorders.28 Current evidence also suggests an association between phthalate exposure and reproductive toxicity, hormonal imbalance, and fetal deformations in humans.31
Drug Cross-Contamination
Eighteen papers32, 33, 34, 35,36 reported cross-contamination with another drug product. One study was a systematic review that covered several cross-contamination cases, reporting eight cases of contamination with hydrochlorothiazide, two with torasemide, and one with triamterene [34]. In the remaining 17 studies, many contaminants were potent prescription-only medications such as antihypertensive drugs including hydrochlorothiazide, olmesartan, and enalapril; anticancer drugs including vincristine; and immune-modulating drugs such as azathioprine.
One key contribution to cross-contamination was the use of shared manufacturing equipment, particularly improper cleaning between the production of different products. Even after proper cleaning, cross-contamination can still occur,33 which highlights areas for improvement in cleaning validation. Another cause identified was human error during production. Personnel shortages and overloaded facilities can result in disorganized equipment and material flow, resulting in mix-ups of products.36
Diuretics such as hydrochlorothiazide have falsely indicted athletes for doping,32, 34 even when the contaminant was present in small amounts. In the cross-contamination of itraconazole with rilmazafone, at least 245 patients reported dizziness, intense drowsiness, and loss of consciousness as well as 2 deaths and 38 traffic accident cases.35 Cross-contamination involving vincristine also led to neurologic symptoms such as weakness and paralysis in at least 107 patients across 12 hospitals in China, despite investigations revealing that vincristine was only present in trace amounts between 0.28 and 18 micrograms per milliliter.33
Where cross-contaminated drugs were not released into the market, drug shortages can still plague consumers and healthcare institutes. In the cross-contamination of Johnson & Johnson’s COVID-19 vaccine with AstraZeneca’s COVID-19 vaccine, up to 75 million doses were ordered to be discarded following the incident,36 and vaccine shortages were subsequently reported in South Africa and Germany.37
Cell Culturing Contaminants
Contaminants associated with the use of cell culturing were widely covered in some studies.38, 39 Such contaminants included bacterial host cells used in the cultivation of recombinant proteins, their endotoxins, and plasmid DNA,[39 as well as tumorigenic stem cells. These studies briefly covered the risks associated with such contaminants, such as immunogenicity,31 but otherwise were more focused on evaluating potential improvements to processes such as identification and purification.
Analysis of Trends by Frequency
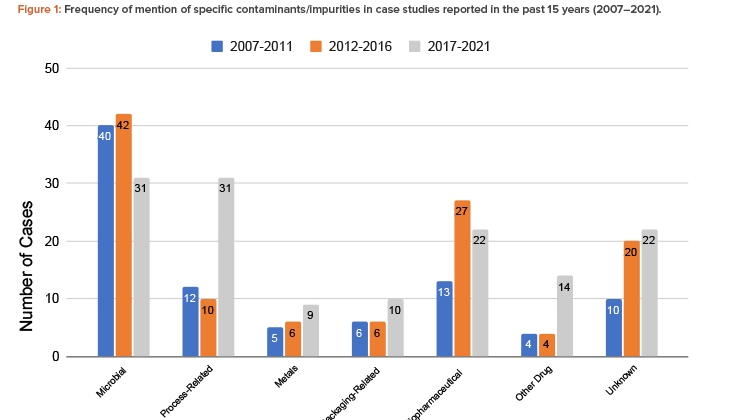
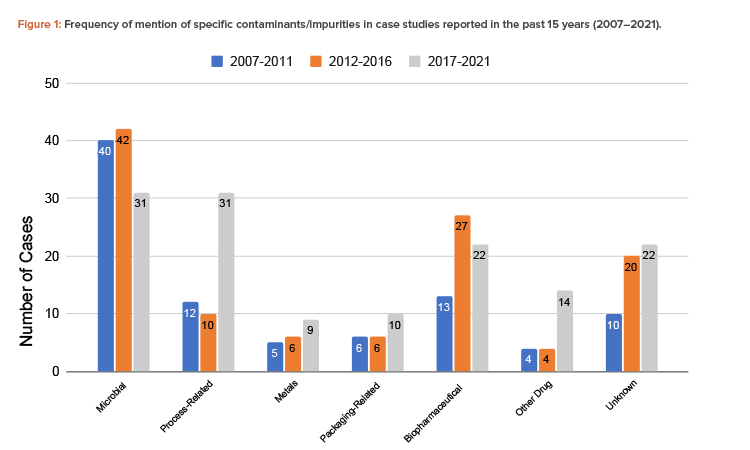
The findings showed that the total number of contamination studies reported over the past 15 years is 344, with 90 in 2007–2011, 115 in 2012–2016, and 139 in 2017–2021 (Figure 1).
Microorganisms are the most common contaminant, followed by biopharmaceutical contaminants and process-related impurities. The number of cases of process-related impurities rose sharply in the 2017–2021 period, due to nitrosamine contamination cases. Notably, aside from biopharmaceutical contaminants, these trends were also observed in the initial study of recall databases of the different RAs. Cross-contamination by other drugs also rose in that same period. The increased number of cases involving these contaminants suggests that closer attention should be paid to the control of cross-contamination and processes involving chemical reactions and the quality of reagents. The importance of segregating production operations in shared facilities should be emphasized. A risk analysis should be carefully conducted when there is any deviation in any of the processes, chemical reactions, and type and quality of the materials, including solvents and reagents.
Countries Commonly Associated with Contaminated Medicinal Products
Due to the globalization of the world today, the issue of contaminated medications is an international one. Any major contamination event that warrants a large-scale recall would likely affect patients globally.2, 40 Although our analysis hinted at the US being most affected by recalls of contaminated medicinal products,5, 29 this could be explained by the fact that the US FDA regularly publishes alerts and recall notifications on their website to communicate recall information to consumers.41
Our analysis also showed that besides the US, the countries of origin where contaminated products have been commonly reported include the UK, Europe, Japan, China, and India.40, 42 The contamination cases appeared disproportionately high for China and India compared to the rest of the world. This observation may not be surprising, given the high production output of these countries where labor costs are lower. Incidentally, the high-profile contamination cases involving nitrosamine-
contaminated drugs and heparin also originated from these countries. It is therefore of interest to compare the GMP standards of WHO, PIC/S, and the previously mentioned major countries to better understand the factors that could have contributed to the contamination events.
Impact of GMP Standards on Contamination
The following components of GMP standards were identified to be pertinent to contamination control: cleaning validation; water quality; sterility testing; buildings, facilities, and equipment; and personnel. The components of GMP standards from WHO, PIC/S, the US FDA, China NMPA, and India CDSCO were analyzed and these GMP standards are as follows:43, 44, 45, 46, 47, 48
- WHO: GMP for Pharmaceutical Products: Main Principles, GMP for Active Pharmaceutical Ingredients, and GMP for Biological Products
- PIC/S: Guide to GMP for Medicinal Products Part I and Part II and Annexes
- US FDA: 21 CFR Part 210 Current GMP in Manufacturing Processing, Packing, or Holding of Drugs and 21 CFR Part 211 Current GMP for Finished Pharmaceuticals
- China NMPA: Good Manufacturing Practice for Drugs
- India CDSCO: Drugs and Cosmetics Act (DCA) Schedule M
The authors’ analysis found that the GMP standards from these agencies are fairly consistent. Most points pertaining to the prevention of contamination are similar in concept, with differences in phrasing and content arrangement. Such differences can still create confusion among manufacturers in relation to contamination control and overall GMP compliance.
In this regard, PIC/S has led the way in publishing a revised Annex 1 to its Guide on GMP for Medicinal Products, which will come into effect 25 August 2023. Annex 1 clarifies the clean air classification and microbial monitoring limits that manufacturers of sterile products have to implement for various processing and sterilization operations—such as aseptic processing, terminal sterilization, and finishing of the sterile products—based on a contamination control strategy and quality risk management principles.45
Another key difference among national and international GMP standards is the level of technical details for cleaning validation. The WHO and PIC/S standards are the most comprehensive, covering changeover between different products, bracketed products, and different batches of the same product. Conversely, national standards of some RAs tend to be devoid of details, leaving discretion to the manufacturers. Improperly validated cleaning procedures for shared production equipment can be a potential source of cross-contaminants, especially during product changeover. Overall, the various GMP standards appear sufficiently comprehensive in terms of contamination control measures. However, the continued occurrence of contamination and cross-contamination events highlights other challenges faced by manufacturers and RAs.
Manufacturer and RA Challenges
Currently there are still differences amongst GMP standards, for example in clean air classification, microbial monitoring limits, cleaning validation, and conditions mandating dedicated drug manufacturing facilities.50 Although manufacturers may abide by the standards adopted by a certain RA, they may be deemed noncompliant to another.
Even when manufacturers abide by the respective GMP standards, there is still a chance, albeit a small one, for contamination to occur. This is due to the impracticality in performing total quality checks for all product items during batch manufacture and characterizing all impurities in a product. Contamination events can still slip through the cracks and defects may only be spotted after release into the market. The increasing use of biopharmaceuticals adds to the complexity of quality control. Additionally, not all manufacturers have the resources to adopt more effective technology to address contamination issues.49
Another major problem can arise from the presence of legally ambiguous gray areas. This is best exemplified in the form of large-scale compounding pharmacies in the US; the FDA has limited power to enforce interventions in compounding pharmacies15 due to ambiguity in whether their activities are considered pharmaceutical manufacturing. Therefore, compounding pharmacies could produce medications in bulk while receiving reduced oversight, leading to various outbreaks of serious contamination.9 This has highlighted the need to assess the presence of possible equivalent gray areas in countries outside of the US. Both China NMPA and India CDSCO face similar issues.46, 47
Further, difficulties may arise when overseas inspections of pharmaceutical manufacturers are initiated. These are most notably observable in terms of the activities carried out by the US FDA, ranging from the need to announce inspections in advance, which gives time for manufacturers to rectify any issues,50 to staffing issues that affect inspection capacity and restrictive policies.51 Collectively, these problems can lead to infrequent overseas inspections. The median inspection frequency between 2011 and 2019 is inadequate and undesirable, standing at approximately two years for high-risk sites and more than three years for low-risk sites.52 Other RAs face similar staffing and labor issues.
Proposed Solutions
Manufacturers
Contamination can be caused by many factors, including personnel’s lack of knowledge and training. The requirement for well-qualified personnel, continual training, and qualification should be strongly emphasized. Manufacturers should embrace a proactive quality culture.
Manufacturers should also be encouraged to harness advanced containment and process analytical technologies, which are already in existence. Manufacturers should be encouraged to harness technology such as quality by design (QbD) when considering problems associated with the final testing of products—from the need to test large numbers of finished products to identify contamination at extremely small percentages to the use of destructive testing—and to place particular emphasis on its practical implementation. A focus on developing and adopting real-time, nondestructive methods of contamination monitoring throughout the manufacturing process is needed, such as by using spectroscopic methods including Raman spectroscopy to improve the speed of contaminant detection.
Contamination issues are a big challenge for compounded medicines. There is a need to reduce the level of human-performed operations, which are a major source of contamination. One possible way to combat this would be to assess which products are most commonly compounded and to create similar formulations to be batch-manufactured, avoiding the need for compounding. Alternatively, the use of robotic compounding and other automated processes could be explored, as these have been shown to reduce contamination rates.53
RAs
Although all GMP standards share a common aim to guide the production of safe and good quality medicinal products, the contents of these national standards are often organized, arranged, or structured differently. These differences may lead to confusion among manufacturers with regard to GMP compliance, including contamination and cross-contamination control. Some GMP standards still use subjective and vague terms such as certain drugs, highly active or highly sensitizing drugs, or cytotoxics, which are left to the manufacturers. It would be best to eliminate these vague terms and to characterize drugs in a globally accepted, common GMP standard to avoid ambiguity.54
A globally harmonized GMP standard for medicinal products in finished dosage forms such as that for the manufacture of active pharmaceutical ingredients (APIs)—namely the PIC/S Guide to GMP for Medicinal Products Part II—can eliminate such ambiguity and confusion. This will go a long way in enhancing overall GMP compliance and quality assurance in the pharmaceutical manufacturing industry. It is also in line with the mission of PIC/S to lead in the international development, implementation and maintenance of harmonized GMP standards.
PIC/S has recently revised Annex 1 of its GMP standard for the manufacture of sterile medicinal products, which goes into effect 25 August 2023. To date, PIC/S has 54 participating authorities and its membership is growing. PIC/S has had success in driving the international harmonization of the GMP standard for APIs. The next challenge is for PIC/S to do likewise for the GMP standard for finished dosage forms, namely the PIC/S Guide to GMP for Medicinal Products Part I.
Through the international harmonization of a common GMP standard, the inspection in large countries such as China, India, and the US can also be more consistent, thereby alleviating the issues of varying inspection standards by local RAs. As outlined in the PIC/S 2023–2027 Master Plan, PIC/S aims to harmonize and standardize GMP training internationally to ensure that its inspectors consistently apply GMP enforcement and inspection to ensure that manufacturers across the world are held up to the same standards regardless of region.
This harmonization also paves the way for mutual recognition agreements and inspection reliance, where any PIC/S member country may recognize the GMP of another PIC/S member country, thus avoiding duplication of inspection which then confers time and cost savings for both manufacturers and RAs. With a harmonized GMP standard, the quality of medicinal products can be assured and be in the best interests of public health. This global cooperation of inspections can also allow for inspections to be done more proactively by eliminating political barriers.
One key issue that remains, however, is the authority granted to inspectors, thereby limiting the routine inspection of overseas manufacturers. As previously noted, US FDA inspectors are not conferred sufficient authority to conduct unannounced overseas inspections, which has contributed to inspections being done infrequently.52 Aside from GMP harmonization, there should also be more authority granted to PIC/S or WHO inspectors to conduct unannounced inspections to assess GMP compliance.
This would avoid incidents where manufacturers that are notified of an upcoming inspection use the lead time to clean the facility and ensure GMP compliance just before inspection,50 giving a false impression to inspectors. These additional inspections may even go further to assure product quality and strict GMP compliance by mandating routine inspections to be conducted at a specified frequency (e.g., at least one inspection every 18 months), to complement the current risk-based inspections.48
Conclusion
This article has investigated the published literature on the contamination of medicinal products to identify common contaminants and contamination trends. It has further analyzed the GMP standards from the WHO, PIC/S, the US FDA, China NMPA, and India CDSCO, as well as the challenges faced by manufacturers and RAs, before proposing possible solutions. Microbial contaminants and process-related impurities were the most common contaminants, with cross-contamination involving other drugs becoming a problem. There are some minor differences among the GMP standards, but they all embody similar concepts regarding contamination prevention.
The main issues for contamination still occurring today could be attributed to lack of knowledge, noncompliance to GMP, confusion due to differing GMP standards, and ineffective enforcement. Possible solutions include the strict requirement of well-trained personnel, continual training, minimization of compounding activities, adoption of QbD and new technology, and GMP harmonization and standardization. PIC/S has led the way in publishing clearer clean air classification and microbial monitoring limits, which manufacturers of sterile products have to implement for various processing and sterilization operations.
It is hoped that the clarifications in the recently updated PIC/S Guide to GMP for Medicinal Products Annex 1 will eliminate existing ambiguities and will eventually result in lower rates of contamination and a higher level of quality assurance for sterile medicinal products. If this happens, international harmonization to the PIC/S Guide to GMP for Medicinal Products, including Annex 1, could be adopted by all RAs and form the basis of international harmonization. It is acknowledged that the contamination cases captured may not be exhaustive, but collectively, they show certain trends have occurred worldwide. It is also acknowledged that the results might have skewed toward countries with greater information availability, despite efforts to include contamination cases globally.
Regardless, the findings have provided a broad overview on the issue of contaminated medicinal products and potential solutions to counter contamination. Future studies surrounding contamination could focus on categorization of common contaminants to aid in QbD and the promotion of shared interests and greater international collaborations.
About the Authors




