

Supplier Qualification Program for Key Raw Materials

With the publication of recent guidance, specifically the US FDA Quality Systems Approach to Pharmaceutical cGMP Regulations
The supplier qualification program is an evaluation of raw material suppliers. The requirements for supplier qualification are wide-ranging and complex, and a qualification process should identify and mitigate the associated supply risks of raw materials and services. Different regulations and guidance for medicinal drug products for human or veterinary use and investigational medicinal drug products must be followed, and various European directives and GMP guidelines also define requirements and expectations.3, 4 For this article, “raw material” is considered any material that is somehow employed in a GMP-regulated process, and “supplier” is used in this discussion as a general term to encompass source manufacturers, vendors, re-packagers, and distributors.
Supplier qualification can also be considered a risk assessment tool because it produces an acceptable level of assurance that suppliers, vendors, and contractors can supply consistent quality of raw materials and services in compliance with applicable requirements. The quality system approach calls for periodic auditing of suppliers either by paper or on-site, and the approval process may include proof or completion of some activities and documentation.
Auditing suppliers is an expensive task that requires a serious commitment of time and resources. However, from a business perspective, it makes good sense to evaluate suppliers at a frequency and level of requirements appropriate to their impact on the final drug product. Several papers have been published about supplier qualification strategies for active pharmaceutical ingredients (APIs), excipients, and primary packaging components.5, 6, 7, 8, 9 This article focuses on non-GMP-regulated raw materials such as detergents, disinfectants, gowning, and other consumables for life sciences applications.
Risk-Based Material Evaluation
A wide range of raw materials can affect product quality or process performance. Raw materials include APIs, process aids, materials, contacting process fluids, excipients, devices, and primary and secondary packaging. Raw materials may be further classified by their use in the manufacturing process and their subsequent effect on quality.
For simplification purposes, in this article, the categories are defined as follows:
- Starting raw materials: These materials are known to significantly affect product quality, are well characterized, and may be part of the medicinal products or in direct contact with them. Examples include APIs, excipients, USP-grade reagents, and primary packaging components.
- Key raw materials: These materials impact process consistency, but do not significantly affect product quality. They may be characterized as thoroughly as needed based on risk. Some examples are detergents, disinfectants, and food-grade lubricants. Also, they may include cleanroom gowning, commodity chemicals, secondary packaging components, and other processing aids.
- Non-starting or non-key raw materials: These materials do not meet the other categories.
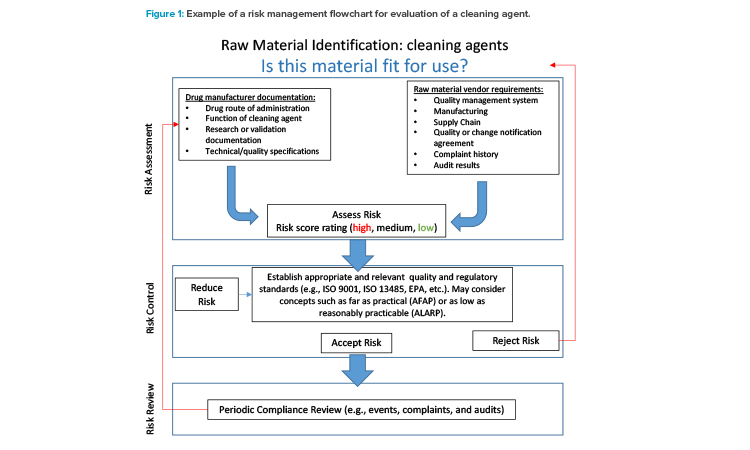
Regulatory guidelines focus on manufacturing practices for the starting raw materials intended to be parts of the medicinal product, such as APIs, excipients, and primary packaging components. The guidelines for starting raw materials define similar GMP requirements for drug products,10, 11, 12 which is reasonable because APIs and excipients are recognized as primary materials for medicinal products, and are therefore a potentially higher risk to final product quality.
Key raw materials used in the facility are considered important due to their role in a validated process (e.g., cleaning validation, sterilization processes, and disinfectant qualification), but they are not regulated by the FDA or any other GMP authorities. Virtually no industry standards have been established for most key raw materials. Further, guidance that specifically addresses supplier qualification has not been formally established, especially for key raw materials, which makes establishing supplier qualification processes even more challenging and reliant upon each company’s requirements.
The supplier auditing program should be based on the risk associated with the material being provided.13 Raw materials should be classified as high, medium, or low risk depending on the criticality of the medicinal product or process. If the pharmaceutical manufacturer has many suppliers, then these suppliers should also be assessed by classifying them into different levels based on their impact on the medicinal product.
The ICH Q9 Quality Risk Management guidelines offers principles and tools applicable to different aspects of pharmaceutical quality.14 As shown in Figure 1, risk assessment becomes a critical aspect in the qualification and management of raw material suppliers. Therefore, the ICH Q9 guideline can be a useful reference when creating a supplier qualification program. The example in Figure 1 relates to cleaning agents used for cleaning validation of processing equipment. The risk management process could be implemented retrospectively for currently used cleaning agents and prospectively during cleaning process development.
The following questions may be used to guide the risk assessment process:
- In what part of the product manufacturing process is the material used?
- What role does the supplier play in the supply chain?
- Has the validation or product development team determined the classification of this raw material? Why or why not?
- Is this supplier the sole source of the raw material?
- To what industries does the supplier provide materials?
The need for supplier qualification may be misinterpreted during the early stages of product or process development, such as clinical trials and revalidation work.15 For example, it is expected that the raw material used in the development phase, not the supplier, will be qualified during stage 1 of the life cycle model, as discussed in the FDA Process Validation Guidance.16 Raw material qualification differs in that the focus is on demonstrating that the material is adequate for the process (e.g., manufacturing, cleaning, and sterilization). However, the raw material supplier will subsequently be qualified should the development or validation groups determine that the material or components will be used in the commercial-scale process. Table 1 is a good example of how the ICH Q9–recommended risk assessment tools can be valuable when evaluating multiple suppliers of the same raw material type.
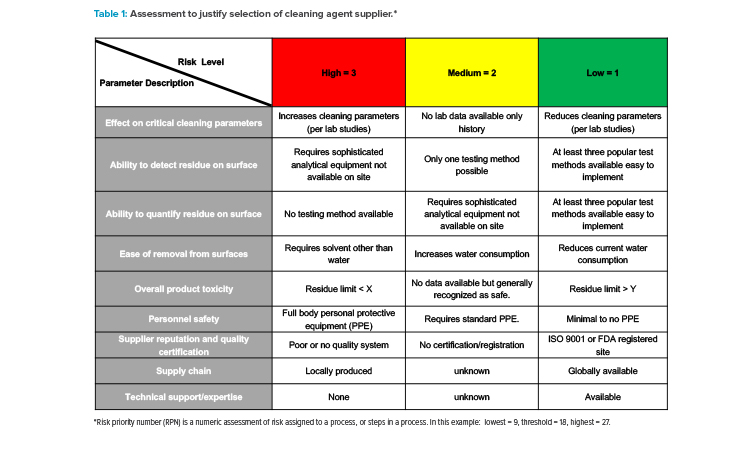
Table 1 depicts the foundations of such a risk assessment to determine the appropriate level of quality and technical requirements by including the two primary principles issued by ICH Q9 : (a) that the evaluation of the risk to quality could be based on scientific knowledge and ultimately link to the protection of the patient, and (b) that the level of effort, formality, and documentation of the quality risk management process could be commensurate with the level of risk.14
Industry Trends
Supplier qualification should be completed before the pharmaceutical manufacturer reviews. The qualification relies on approval of the test results reported on the certificate of analysis or conformance and on at least one on-site identity test.
The general supplier approval procedure for key raw materials starts with the buyer, purchasing, or procurement department contacting the preselected supplier. An internal specification sheet is created and sent to the supplier for review and approval. Supplier assessment surveys, also known as paper audits, may also be sent to the supplier at this point. The supplier-completed questionnaire is then received by the company’s procurement and then quality departments. Suppliers may be required to provide samples (if first qualified), and the quality control lab tests those samples. The samples provided will be analyzed by quality control (QC) and reported to quality assurance (QA). If the results comply with the established specification, then QA may plan for an on-site supplier audit. An on-site supplier audit is planned and scheduled. If the on-site audit results are satisfactory, then the supplier is considered approved.
It is important to note that all steps mentioned may not apply to all key raw materials and may vary per company. As previously mentioned, the supplier qualification requirement should consider the risk classification of the material. Over the years, global companies have established minimum sup-plier qualification requirements including quality surveys, quality agreements, on-site audits, and technical support.
Quality Surveys
To determine if a supplier can meet expected quality requirements when supplying raw materials, a questionnaire may be used to gain information about the quality standards, regulations, certifications, or best practices applicable to the type of key raw material being supplied. Surveys should contain questions applicable to the approval of a particular supplier. While it is important to know that a supplier of key raw materials has appropriate quality systems and best practices while manufacturing key raw materials, the materials are not GMP regulated, and full adherence to the GMP regulations established for drugs, medical devices, or other GMP-regulated materials is not realistic. The best that can be expected is a key raw material being manufactured “at an FDA registered site” or “manufactured under a quality system that models a GMP-compliant quality system.”
Quality surveys are intended to provide a basic understanding of the supplier’s quality management system. Questions should be straight to the point and clear, and companies should be cautious about including questions unrelated to quality systems such as pricing, environmental health and safety practices, or product technical questions. Instead, other survey forms that focus on those business aspects can be sent separately. Because of proprietary and company confidential restrictions, many key raw suppliers may omit details when responding to survey questions that ask to “describe,” “explain,” or “attach a copy.” For the same reasons, a supplier may deny some information (e.g., highest education level achieved by an individual in a certain position) if irrelevant to quality systems.
Quality Agreements
In November 2016, the FDA published the guidance Contract Manufacturing Arrangements for Drugs: Quality Agreements, which describes the agency’s current expectations for firms that outsource the production of drugs subject to current GMP regulations. 17 This guidance has been the basis for quality agreements in the industry, even though it is focused on contract manufacturers instead of raw material suppliers. Nevertheless, the concepts in the guidance document could be applied in the quality agreement to establish the expectations between the contract giver (company) and contract acceptor (supplier). Several important aspects for quality agreements are discussed or recommended in the literature.18, 19, 20, 21
The following aspects must be clearly stated and agreed upon:
- The roles and responsibilities of the company and the supplier
- How deviations and out-of-specification results will be investigated, documented, and resolved
- How changes that may need to be made to the manufacturing process, equipment, analytical methods, or specifications are managed and communicated
- How complaints are handled and resolved
- What rights the company has for on-site audits and management of audit observations
Common issues with quality agreements about key raw materials are that they often prohibit all changes without first obtaining the company’s consent. First, this type of broad prohibition exceeds the legal requirements applicable to medicinal drugs, which permit routine, non-major changes to be made without first notifying the FDA. By unduly restricting non-major process improvements, companies may substantially undermine the suppliers’ ability to implement quality-improving, efficiency-generating, and cost-saving measures that, in the long run, benefit both parties.
Additionally, it is not logistically possible for suppliers of non-customized globally available key raw materials to contact every end user and request consent to proceed with a change. For example, if a key raw material supplier accepts a contract with excessive change notification requirements without review, this could eventually compromise the supplier’s ability to maintain compliance with the established quality agreement between both parties. On the other hand, suppliers must acknowledge the needs of GMP-regulated companies and avoid significant changes that affect product quality, fit, form, and function, which may impact the use of the key raw material by companies in validated manufacturing. When unavoidable, all efforts should be made to ensure that the company is notified in a timely fashion and provided sufficient information and product supply to address their validation concerns.
| Compliance Topic* | Reference Guidance |
Pertinent Question(s) for Supplier |
|---|---|---|
| Transmissible spongiform encephalopathies (TSE) and bovine spongiform encephalopathy (BSE) |
27, 28 | Is the material produced with animal-derived ingredients? If so, what can you tell us about the animal derived ingredient(s)? |
| Residual solvents | 24 | Is the material produced with class 1, 2, or 3 solvents? If so, what can you tell us about the solvent ingredient(s)? |
| Elementary impurities | 25, 26 | Is the material tested for elementary impurities? If so, what can you tell us about the impurity(ies)? Or Are metals or metal catalysts used to produce the material? If so, what can you tell us about the metal ingredient(s)? |
| Pallet treatment | 29, 30, 31 | What type of pallet is used to ship the materials: plastic or wood? If wood pallets, are they chemically or heat-treated? |
| Nitrosamines | 32, 33 | Is the material produced with any known N-nitrosamine or nitrosating agents? |
| Melamine | 34 | Is melamine used to produce the material? Or Is the material produced with components that are at risk for melamine contamination? |
| Jatropha | 35 | Is the material produced with components derived from jatropha plant? |
| Phthalates | 36 | Is the material produced with dibutyl phthalate (DBP) or di(2- ethylhexyl) phthalate (DEHP)? |
*This is not an all-inclusive list.
Quality agreements vary in their level of procedural specificity, and often the requirements are inconsistent with the supplier’s standard procedures. Some quality agreements may merely state that the supplier “has procedures” governing a particular area. Other companies may set forth detailed procedures that the supplier must implement for a particular area and these detailed requirements may create issues for key raw material suppliers. For example, the quality agreement may provide a three-year retention period for batch records, but the supplier’s normal procedure may call for a two-year retention period. In this example, although there may be nothing inherently unreasonable about retaining batch records for an additional year, the supplier may want to follow current policies instead of assuming the long-term cost of tailoring its procedures to accommodate a single customer.
On-Site Audits
Pharmaceutical manufacturers are responsible for auditing high- and moderate-risk suppliers, and these audits should be determined on a case-by-case basis. Where an audit is not deemed necessary, this should be justified appropriately, including with a formal risk assessment. When a supplier audit is indicated, it should be conducted by staff with adequate knowledge and training. A written plan for the audit should be prepared before the audit. After the audit, an audit report should record what was reviewed and any observations identified. The supplier should be expected to deliver a written response to any deficiencies, and these responses should be reviewed before the audit is closed. The resulting audit report can form the basis for the approval of the supplier.
The supplier should be re-audited at a specified frequency to verify ongoing performance. A rationale for the minimum audit frequencies for each supplier should be documented. The standard industry practice is every 3–5 years for non-GMP-regulated key raw materials. Even if the initial audit was on site, a desktop and/or questionnaire audit might be acceptable for re-audits if there have been no quality issues and the supplier has a good quality and compliance history.
The COVID-19 pandemic resulted in governments imposing temporary measures such as confinement, quarantine orders, and travel restrictions that are impacting GMP manufacturers in their capacities to perform on-site supplier inspections. Consequently, many drug manufacturers have adopted temporary measures such as performing virtual supplier audits to maintain compliance and supply of medicines to patients. The term “virtual audit” applies to inspections performed off-site using enhanced communication and information technology to fulfill a legal requirement of an on-site inspection. The only difference is that the inspector is not physically present. These audits may also be described as “remote” or as “distant inspections.”
Technical Support
The supplier’s ability to provide technical support is critical for the design, qualification, and monitoring stages of the process life cycle approach. For example, for cleaning agents used in validated cleaning applications, technical support could include laboratory testing for selecting the best cleaning agent and cleaning parameters, which saves time and resources during start-up or when trouble-shooting existing cleaning issues. Technical support should be available via phone calls, emails, teleconferences, webinars, and on-site support if needed. Technical literature may include the following, as applicable: material safety data sheet, certificate of manufacture/analysis, technical data sheets, technical tips, and laboratory reports.
Common Issues With Guidance Documents
A series of supply chain disasters—such as heparin, melamine, and nitrosamines contamination—has resulted in more pressure than ever for pharmaceutical manufacturers to develop better supplier qualification practices.22 Material management and supplier evaluation are key processes to avoid batch failures and adverse effects on patients. As a result, pharmaceutical manufacturers are demanding quality system compliance with adequate standards and increased information transparency from their suppliers.23 Some raw material suppliers require more provenance information from their suppliers, such as source, origin, and other essential information for traceability purposes.
Most FDA (or equivalent agency) guidance documents related to the subjects mentioned previously are applicable to medicinal products and their starting raw materials. However, key raw materials that are not purposely added to or in direct contact with the medicinal product may be beyond the scope of those documents. For that reason, requesting suppliers of key raw materials to make the product fully compliant with such guidance documents is not realistic. In some cases, compliance may not even be feasible due to the type of material.
The USP <467> Residual Solvents and USP <232> Elemental Impurities guidances are good examples to illustrate this issue. The first is a standard for the testing and potential reporting of residual solvents in pharmaceutical products. Residual solvent is defined as organic volatile chemicals that are used or produced in the manufacture of drug substances, excipients, or in the preparation of drug products.24 Similarly, elemental impurities specify limits for the number of elemental impurities in drug products.25, 26 These impurities include catalysts and environmental contaminants that may be present in drug substances, excipients, or drug products. These impurities may occur naturally, be added intentionally, or be introduced inadvertently.
These USP documents do not apply to key raw materials such as cleaning and germicidal agents used in drug manufacturing facilities because these types of items are intended to clean and disinfect surfaces. Some surfaces on which these cleaning agents are applied may also be in direct contact with drug products; however, residues are generally removed before the equipment is used. An effective and validated cleaning procedure will ensure that any potential for residuals from cleaning agents is not transferred over from the cleaning process into the next batch of drug product.
Even though key raw materials may be excluded from USP <467>, USP <232>, and other similar guidance documents, assessing the risk for potential contamination into the manufacturing process is still recommended. A better approach is to ask suppliers more pertinent questions as applicable to the material instead of requesting a declaration of compliance with these standards or guidance documents. Table 2 provides a list of common compliance topics and reference guidance documents with a suggested question for non-GMP-regulated key raw material suppliers.
Conclusion
Considering the regulatory challenges, it is important to have a deep understanding of key raw material suppliers when sourcing materials worldwide. Suppliers must be willing to provide the information needed for regulatory filings or other regulatory requirements, including materials not governed by GMP regulations. Favoring suppliers that can supply reliable and high-quality products ensures safe and effective drugs and makes good business sense.
About the Author



