Software as a Medical Device Fundamentals
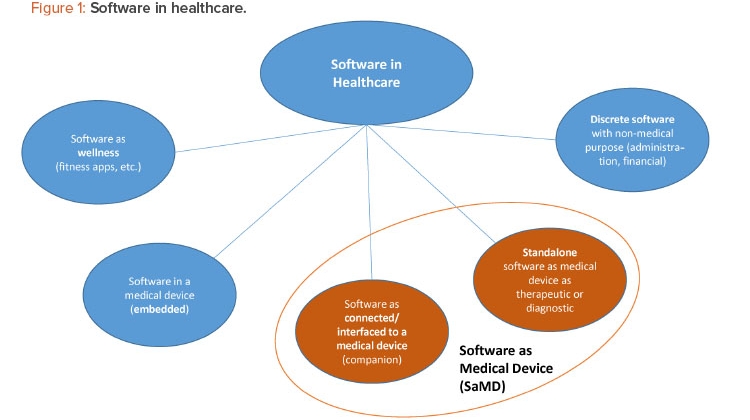
Software as a medical device (SaMD) is software intended to be used for one or more medical purposes without being part of a medical device.
SaMD is one of many medical and nonmedical uses of software in healthcare (Figure 1). For regulatory purposes, SaMD products should not be mixed up with (a) wellness apps or (b) software with a medical purpose that is embedded in a medical device. There are many connected devices and apps that can be used exclusively for wellness or general well-being purposes, such as weight management, physical fitness, sleep tracking, and stress management. Given that these types of devices and apps pose little risk to consumers, the US FDA exempts them from premarket review.3 However, if an app includes medical-related functionalities, such as analysis of data from a sensor for physiological monitoring for a medical purpose that poses moderate to high risk, the app is regulated as a medical device subject to a higher risk classification and premarket review. (SaMD regulation is discussed in greater detail later in this article.)
Regulators exclude software with a medical purpose that is embedded in a medical device from the definition of SaMD because the embedded software (e.g., insulin pump software controlling pump functionality and managing data) is an integral part of the hardware/medical device.4 In contrast, an SaMD typically runs on a nonmedical computing platform, either directly on a smartphone or on remote servers running in the cloud.
SaMD products fall into two main categories:1
- Software connected to/interfaced with a medical device
- Standalone SaMD
Examples of connected/interfaced SaMD products are companion apps with scheduling, monitoring, and sharing functionalities, which are connected via Bluetooth Low Energy to drug delivery devices.4 Standalone SaMD products are used to assist in the treatment of substance use, schizophrenia, amblyopia 5 and stereopsis vision disorders,6 as well as for diagnosis (e.g., for ophthalmic disease progression).7
For pharmaceutical companies, SaMD products are attractive opportunities to offer additional services to patients and caregivers, such as tools for educating patients and managing diseases and tracking symptoms and treatments. Companies can take advantage of many new technologies, such as innovations in imaging processing, data mining, artificial intelligence, and machine learning, to develop digital products that complement their existing portfolios and development pipelines.
For example, SaMD products may help patients adhere to their drug regimens, build good habits, and manage symptoms actively and remotely. The software or app may even offer features that help physicians diagnose a disease or monitor disease progression.
Additionally, digital therapeutics can strengthen or reinforce other forms of therapy. For example, a digital therapeutic currently in development for amblyopia utilizes gaming to train the eyes.5
Data Collection Benefits and Risks
With the consent of the patient, SaMD can be used to collect data, either inside or outside of a clinical setting. SaMD tools may be developed to track patients’ medication use or adherence, or to monitor disease progression or therapeutic effect. Pharmaceutical companies, healthcare providers, researchers, and regulators can analyze such data to learn more about how patients use particular drug products, how often they are prescribed, or what habits patients have in relation to the use of the drug product.
This type of data collection can be a source of information that is different or less expensive to capture than data from a clinical study or focus group. Additionally, SaMD data may be collected from a significantly larger population or sample size than those studied in traditional clinical studies. For instance, an app associated with a diabetes medication can give the patient an opportunity to track weight, blood sugar, or dietary changes.8 The collected data can be used to improve therapies by identifying preferences of patients or healthcare professionals. When a diagnostic tool is based on machine learning, continuous data collection can be used to validate assumptions and collect further data to improve later versions of the products.
Although data collection can be an opportunity for pharmaceutical companies, it is also a risk. Companies must comply with various rules and regulations regarding storage, privacy, and use of data, which can differ significantly from country to country. User agreements and consent forms must include all ways the company wishes to use the data, and the user must be informed of and give consent for how the data will be collected and used. In many cases, user anonymity must be guaranteed, as this is required by regulation and expected by consumers and patients. Finally, data must be stored and accessed in a secure way, and protective measures against cybersecurity attacks must be taken. Pharmaceutical companies are experienced in the collection and protection of clinical study data, but the rules and regulations for data collection and storage activities related to data collection via software may be unfamiliar territory.
Development Prerequisites
Before embarking on SaMD development, companies should carefully consider the resources needed, the timelines, how the organization will need to adapt its processes and culture, and the potential return on investment.
SaMD involves many activities outside of the core competencies of pharmaceutical companies, and this contributes to the risks and challenges of offering digital products. For example, although a pharmaceutical company will likely have a large IT organization and experts in GxP and computerized system validation, managing SaMD products requires different types of IT expertise, which will need to be built within the company and may also depend on help from external vendors.
Similarly, medical device knowledge is typically limited to the relatively small part of the company that develops, tests, and registers medical devices, and the expertise of those working with medical devices may itself be limited to conventional devices for delivery of pharmaceuticals (e.g., single integral combination products, such as prefilled syringes), which may not be considered medical devices from a regulatory perspective. As a result, some companies may not have any employees with sufficient knowledge of the regulatory pathways of medical devices, especially SaMD products.
The phases in the product development and design control process for SaMD products are the same as those for other medical devices, but the phases are often compressed and combined in unaccustomed ways that require specialized expertise and organizational flexibility. For instance, the design and development phase of an SaMD application may make use of agile project management methodologies that combine the development and verification phases, and these kinds of software tests can be performed in a matter of hours (and repeated in case of changes during the development). Also, in contrast to the relatively linear development processes of drug products and other medical devices, SaMD development is a process that continues over the life cycle of the product, with repeated development loops aiming to continuously improve the product.
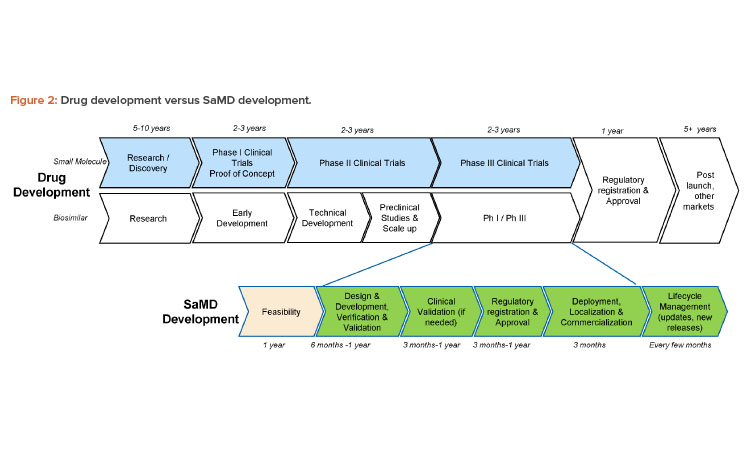
As a result, the timelines for developing digital products can be in the range of months or a couple of years, which means the time to market for an SaMD product is likely to be much shorter than the decades-long effort that may be needed to develop a new drug product in combination with a “standard” mechanical device such as an inhaler (Figure 2). All parts of the organization, including the marketing and commercial divisions, may need to learn to adapt to the shorter timelines of SaMD development and ongoing update/release cycles.
On the other hand, the organization may have unrealistic expectations of extremely short timelines, similar to those for nonmedical apps. The time to market for an SaMD app is longer than for a wellness app because SaMD products are subject to regulatory requirements and must undergo regulatory submissions that can take months, especially if the SaMD product involves novel technologies. (A de novo submission to the FDA may be expected to take a year to complete.) Additionally, compared to wellness apps, SaMD products usually take longer to develop because they involve greater risks, including security risks.
Large pharmaceutical companies have very complex business processes and governance procedures. These are well suited for the risks and payoffs of developing drugs, but they may not be a good fit for an agile and efficient software life-cycle management process, which requires frequent software releases and updates. Organizations will therefore need a business case and value proposition comparatively early in the development of SaMD products to ensure that the products can be designed to meet the needs and requirements of the patients, healthcare providers, and payers.
As the business case is built, the company must recognize that although SaMD products show great promise in general, their value proposition is still not proven. While SaMD products cost much less to produce than new drug products, they will likely generate only a fraction of the revenue of a typical drug product. This discrepancy can make it difficult to fit digital projects into a company’s overall strategy. It can also be easy to underestimate the real costs (including life-cycle management) of these types of projects, especially when there is a learning curve for the company. The payoff will not be instantaneous.
To guarantee a quicker path to digital product realization, good planning, clearly defined requirements/needs, and stakeholder management are essential. First, it is necessary to identify stakeholders early in the project. Whereas branding, labeling, and packaging typically come into later phases of a project, the visual nature and importance of language in most SaMD products requires that these stakeholders become involved earlier in the process.
The various user needs and requirements of patients and healthcare providers should be identified as early in the project as possible, to ensure clarity and avoid delays in the project. Language, country-related treatment differences, and even cultural requirements must also be identified before work on user interfaces is begun.
Relevant regulations should also be identified before work begins. These can include specific requirements for medical devices, SaMD apps, and data collection, use, storage, and security (e.g., the EU’s General Data Protection Regulation).9
Questions about the design of the SaMD product, such as which operating system or platform (smartphone, tablet, or web service) will be used, should also be clarified in the early stages of development.
The company must also be aware that vendors used for SaMD fall under the scope of the vendor management system, and the vendor qualification must be carried out as for any medical device. It is not unusual for a single project to use multiple vendors—for instance, one for the user interface, one for the back end, and another for cybersecurity testing—as well as various consultants. Vendor management, including the management of inter-vendor communication, can add significant complexity to a project; however, choosing the right vendors for the components of the software will also help ensure the success of the project.
Design validation of the SaMD product is another area that may pose challenges for pharmaceutical companies. As with other medical devices, the design validation must show that product fulfills the user needs. Many of these user needs can be assessed by performing human factors evaluations to ensure that ergonomic and usability requirements are met in the final product.
Clinical evidence is also necessary in validation to show that desired clinical outcomes can be achieved. Clinical studies for SaMD products may be a challenge for a pharmaceutical company used to performing Phase I, Phase II, and Phase III studies. Although the methods for SaMD clinical studies are the same as those for other medical devices, SaMD studies may be somewhat smaller, depending on the product’s level of risk. However, companies need to be aware that EU and US regulations for SaMD clinical data are becoming more complex, and the types of clinical data to be collected for a given SaMD product depend highly on the product’s purpose and risk classification.
Deployment and Life Cycle Management
The life cycle of an SaMD product differs to some extent from the life cycle of a classic medical device, and significantly from the life cycle of a drug product. Once a drug product is approved, it typically undergoes as few changes as possible, since a change often necessitates filing an amendment with the regulatory authorities and possibly requires new clinical data. In contrast, software must be updated continuously to keep up with operating system updates, security concerns, and ever-changing hardware. Also, software updates provide opportunities for the developers to make changes based on customer feedback and add new features to stay competitive.
Although software updates are necessary, making frequent updates to SaMD products can be difficult because medical devices have stringent change control requirements—any change in a design or process must be verified and validated unless it can be shown to have no impact on the function and safety of the device. Depending on the organization, the change control process can last weeks to months.
Pharmaceutical companies may need to adapt their processes for handling complaints from patients and healthcare providers to address SaMD-related complaints. Depending on the software design, it may be easier for a user to register a complaint, which may lead to an increase in the frequency or volume of complaints. The distributor and/or developer of software is required to investigate complaints and report any adverse events to the health authorities within a defined time frame.10, 11 Since updates are released frequently, the company must be prepared to investigate (and report, if required) potentially large numbers of complaints within the time frame required by internal company processes and health authorities. The complaints may also be received directly by a vendor or partner, if they are hosting the software; therefore, a robust quality agreement detailing the various responsibilities of all parties in the complaint investigation and reporting is essential for an effective product vigilance process.
To effectively manage the demands of SaMD life-cycle management, the company may need to establish a so-called DevOps organization to react quickly to customer complaints or bugs and promptly implement changes and product improvements. This most likely will involve a flexible, cooperative arrangement with vendors, and reactive product vigilance and complaint-handling processes.
To expedite certain activities, the DevOps organization may operate outside of the normal business processes used for nonconnected medical devices or drug products. For instance, the change control process may allow software vendors to make certain types of security-critical updates without the approval of all departments of the pharmaceutical company, as long as the updates do not affect any function-critical or regulatory aspects. To maximize its efficiency, the change control process must have clear criteria for which departments should be involved in particular types of change. For instance, many types of change may not require regulatory reporting, so the criteria for involving the regulatory team must be clear. In some companies, the DevOps organization may have a streamlined structure, including a specialized regulatory department to assist in reporting any regulatory-critical changes to ensure that the essential functions are available for life-cycle management.
During contractual negotiations with third parties providing software, companies must make sure to address service management. This includes not only the provisioning of services in a normal situation but also setting up protocols to manage potential service disruption scenarios. These protocols should be designed to mitigate any inconvenience to the pharma company or the users of the software, including the transfer of services to a third party where possible. This ensures that contingencies are in place for managing to the extent possible the potential risks that data used for a specific application will be lost or inaccessible.
There are currently a number of standards for SaMD deployment; the most important is IEC 62304: Medical Device Software–Software Life Cycle Processes.12 Additionally, the ISPE GAMP® Good Practice Guide: A Risk-Based Approach to Regulated Mobile Applications13 provides information specific to mobile phone apps, including guidance on when to retire a mobile app, data privacy, and issues related to connectivity.
Regulatory Considerations
There are two types of regulated software for medical devices:
- Software integral to a medical device (software in a medical device)
- Software as a medical device
Table 1 summarizes how the US FDA and the EU currently classify regulated medical device software according to their level of risk. Note that as many new types of software, such as clinical support software, as well as new technologies, such as artificial intelligence and machine learning, are being developed, regulations are developing concurrently. For instance, the FDA has issued a draft guidance on clinical support software.14
FDA
Over the past decade, the FDA has developed a risk-based approach to regulating SaMD while aligning its regulatory approach with the evolving nature of digital devices. FDA regulations vary by risk classification and aim to set the level of control required to ensure a safe and effective medical device:
- Class I SaMD products are subject to general controls, including manufacturer establishment registration and device listing, but most are not subject to a review process prior to being placed into US commerce.
- Class II SaMD products are subject to general and special controls and 510(k) premarket notification marketing clearance.
- Class III SaMD products are subject to the premarket approval application process and must be supported by clinical evidence of safety and effectiveness.
| US FDA | EU |
|---|---|
| Class I: low risk • Example: otoscope mobile application |
Class I: low risk Example: fertility tracker |
| Class II: moderate risk • Example: mobile app to monitor physiological processes |
Class IIa: low–medium risk • Example: mobile app to monitor physiological processes that are not considered to be vital Class IIb: medium-high risk • Example: mobile app intended to analyze a user’s heartbeat, detect abnormalities |
| Class III: high risk • Example: Closed-loop application (e.g., artificial pancreas) |
Class III: high risk • Example: Closed-loop application |
For more information on the FDA approach to regulating medical device software, refer to the agency’s “Policy for Device Software Functions and Mobile Medical Applications” 15 and its webpage dedicated to SaMD.16
EU
In the European Union, medical devices are regulated under the Medical Devices Directive (93/42/EEC)17 and the In vitro Diagnostics Directive (98/79/EEC).18 Pursuant to the directives, standalone software has a “medical purpose” if it is intended by the manufacturer to be used for humans for the purposes of
(a) diagnosis, prevention, monitoring, treatment, or alleviation of disease; (b) diagnosis, monitoring, treatment, or alleviation of, or compensation for, an injury or handicap; (c) investigation, replacement, or modification of the anatomy or of a physiological process; or (d) control of conception.
Under the 93/42/EEC directive,17 the conformity assessment for a Class I device is performed by the medical device manufacturer. A certified Notified Body (NB) assessment of conformity is required for devices in Classes IIa, IIb, and III.
Under the 98/79/EC directive,18 in vitro diagnostic devices are classified into four categories: general in vitro diagnostics, self-testing in vitro diagnostics, List A, and List B. A general in vitro diagnostic device is self-certified by the manufacturer. NB assessment of technical documentation is required for self-testing, List A, and List B in vitro diagnostic devices.
Guidance about the classification of standalone healthcare software is offered in the European Commission’s “Guidelines on the Qualification and Classification of Stand Alone Software Used in Healthcare Within the Regulatory Framework of Medical devices”.19. In these guidelines, the criterion for medical device classification is whether the software is intended to interpret or facilitate the interpretation of data by modifying or representing health-related individual information.
In 2019, the European Working Group on Borderline and Classification issued an updated version of the “Manual on Borderline and Classification in the Community Regulatory Framework for Medical Devices”, 20 which provides guidance for cases in which the classification of a device as medical is not straightforward.
In 2017, the European Commission promulgated the Medical Device Regulation (MDR) 2017/745,11 which goes into effect in 2021, and the In Vitro Diagnostics Regulation (IVDR),21 which goes into effect in 2024. Under the MDR, SaMD is classified under Rule 11 based on the level of risk associated with its use. The MDR also defines SaMD as software that drives a device or influences the use of a device. Further, the MDR specifically exempts software intended for general purposes, even when used in a healthcare setting, and states that software intended for lifestyle and well-being purposes is not a medical device.
In response to the COVID-19 pandemic, the European Commission postponed the application date for the MDR to 26 May 2021.
In October 2019, the Medical Device Consulting Group released “Guidance on Qualification and Classification of Software in Regulation (EU) 2017/745–MDR and Regulation (EU) 2017/746–IVDR”.22 The document defines the criteria for the qualification of software that falls within the scope of the new medical device regulations and provides guidance on the application of classification criteria for software.
Case Study
The following case study of an inhaler and drug product packaged with a sensor highlights challenges that pharmaceutical companies face when introducing SaMD products to the market.
The Business Case
A major reason why asthma patients and patients with chronic obstructive pulmonary disease fail to respond to treatment is poor compliance to prescribed treatment programs. In particular, suboptimal adherence to asthma treatment can affect more than 50% of patients prescribed such treatment.23

To address this problem of poor treatment compliance, Novartis initiated a partnership with Propeller Health, a digital health company that develops and manufactures a variety of sensors that track when the patient takes a dose and communicate medication use to the app on the patient’s smartphone. These sensors can attach to various types of inhalers, including dry powder inhalers commonly used for maintenance treatment as well as pressurized metered dose inhalers commonly used for acute rescue treatment of exacerbations with short-acting beta antagonists. Using Novartis-developed acoustic technology, Propeller developed a sensor that can be attached to a Novartis inhaler, enabling patients to use the Propeller digital health app to manage their condition in partnership with their healthcare providers.
Novartis and Propeller initially partnered to use the sensor in clinical trials to record when patients were taking their doses, as the recording of medication use by a sensor was expected to be more reliable than self-reporting. However, Novartis saw the drug-device combination as a way to distinguish its inhalation products from other inhaled therapies expected to reach the market at nearly the same time. Thus, the business case quickly evolved into an opportunity to co-package the sensor with a drug-device combination product, creating an SaMD-enabled product that can motivate patients to take their maintenance medication regularly by providing reminders to patients while tracking and trending their inhaler use.
Product Features
The sensor is provided to patients as a system pack, which contains an inhaler device, the sensor (shown in Figure 3), and app access, along with the first 30-day supply of the drug product, in the form of capsules for inhalation. For the subsequent months, the patient can be prescribed a typical inhaler/drug patient kit, which includes a 30-day supply of medication along with the inhaler. The patient will remove the sensor from the used inhaler and attach it to the fresh inhaler. The sensor can be used for up to one year, and then the patient can be prescribed a new system pack.
The sensor itself has several functions:
- It senses when the patient takes a dose.
- It reminds the patient when it is time to take a dose.
- It communicates with the smartphone app to show various dosing trends, dose reminders, and other useful information for people with asthma.
The associated smartphone app is an example of SaMD interfaced to a medical device. Its features, in addition to the sensor, are:
- It allows patients to review their asthma medication regimen for the day and receive useful information about their disease, weather, and other triggers.
- It provides information about the patient’s weekly or monthly medication use.
- It allows patients to view details for each use of their medication, add rescue inhaler use events, and record symptoms.
- It contains reports that can be shared with a healthcare provider, information on prescribed inhalation therapies with specifics on when dose reminders are set, and other information related to the sensor itself (e.g., sensor attachment and removal instructions).
- It enables patients to add other medications (such as rescue treatment) for manual tracking.
Data from the sensor also creates a web-based report about the patient’s use habits of their therapy, which healthcare providers can access via email (with patient consent) or discuss with the patient during appointments. This report can help the HCP work more closely with the patient, make better-informed therapeutic decisions, and give appropriate advice to improve patient adherence to the treatment regimen.
Regulatory Approval
The sensor variants currently have FDA 510k approvals and are distributed within the US. In anticipation of entering the European market, the sensor underwent a conformity assessment to obtain the required Conformité Européenne (CE) mark, which demonstrates that the sensor meets the legal requirements to ensure it is safe and performs as intended, as required previously under the EU’s MDD, and under the MDR when required.
The companies believed that healthcare providers would appreciate the convenience of prescribing a kit containing the inhaler and medicinal product as well as a sensor in the same package. Therefore, they decided to include the system pack (containing the inhaler, drug product, and sensor) in the submission for the drug product.
Among the many challenges in bringing the system pack to market was determining the precise role that Novartis would play in the distribution of the sensor. By supplying the sensor as a part of the system pack, Novartis took on the role of a “system assembler” under the EU regulations (MDD and MDR). Under the MDR, a system assembler is considered to play the economic roles of manufacturer and distributor. Because of this, Novartis needed to show that the sensor was appropriate to use with the inhaler and drug product. A full medical device development project was initiated for this system pack, with user needs, design inputs and outputs, risk management, verification testing, and validation testing, which included a human factors summative study.
This development process was risk based and consisted of assessing the interface/interoperability of the sensor and app with the user, inhaler, and drug product as a supplement to the existing technical documentation of the system. For instance, the use-related risks included setting up the system incorrectly and the effects of getting dose reminders from the sensor and/or app. Additionally, system risks were assessed for any potential effect that the sensor could have on the pharmaceutical performance of the inhaler (e.g., due to the sensor’s electronics or by limiting inhaler functionality). This all had to be completed under ambitious timelines, before the scheduled drug product submission to EMA.
As a system assembler, Novartis had to ensure that the app content was aligned for EMA regulations. Although the sensor is CE marked, it did not undergo EMA approval, and the app content is considered by EMA as drug product labeling because it instructs users on how to administer the drug. To ensure that the initial version of the app as well as any updates conformed to EU regulations, a review process was developed, which included the coun-try pharmaceuticals organizations because the app is available in many languages. This review process assessed, among other topics, the app content related to dosing. For instance, the app was required to instruct patients to take exactly one dose daily, as is instructed by the patient leaflet. Also, the app had to avoid any implication of direct-to-consumer marketing, which is forbidden in the EU.
Finally, the quality agreement between Novartis and Propeller had to define the various roles of both companies in the event of a customer complaint, as either partner could potentially receive complaints for the other’s product. This agreement had to include the specific responsibilities of each party to pass on any information to the respective manufacturer of the components. For instance, Propeller, as the developer of the app, is likely to be the first point of contact for any customer questions or complaints; however, Novartis is responsible for the drug product safety. Therefore, if Propeller receives a complaint about the drug product, they are required by the quality agreement (as well as European regulation) to pass on this information within a specific time frame so that Novartis can investigate if an adverse event has occurred.
Despite all of these challenges, the project was a success. The joint EMA approval of the drug product, along with the system pack, was the first digital approval in Europe of a sensor with an inhalation product.
Conclusion
SaMD products are becoming increasingly important user-centric products for the pharma industry. A clear understanding of the SaMD risk categorizations and regulations is essential when determining the development and life-cycle management effort. As described in this article, SaMD products, especially when connected to sensors and drug delivery devices, have the potential to substantially improve therapy outcomes (compliance, training, behavior change, etc.) compared to what has been possible with conventional medical devices.
About the Authors



