Opportunities in Continuous Manufacturing of Large Molecules

Continuous manufacturing has attracted significant interest over the past decade for small molecules formulated as drug products. The case for adopting continuous manufacturing platforms for manufacturing biologics (i.e., large proteins or biologic products such as vaccines) would, in principle, be even more justified for both quality and business gains. This article briefly reviews continuous biomanufacturing (CBM) at a time of very high and global demand for vaccines as well as increased demand for cell and gene therapy products.
Biologics are very large molecules, complex to produce, with stringent aspects on interchangeability. Therefore, compared with small molecules, they present a considerably bigger challenge and have higher criticality in terms of manufacturing sciences and technologies, availability to patients, and the regulatory processes involved.
In June 2020, ISPE hosted the Continuous Manufacturing Virtual Workshop, a meeting where continuous manufacturing for both small and large molecules as drug substances and formulated drug products was addressed.1. For a broader perspective on emerging continuous and integrated platforms for recombinant proteins, a review of novel technologies to enable continuous manufacturing of biologics, and specific analytics considerations for the reader is referred to the online presentations from that workshop,1 as well as to “Biotech Processes: Challenges for Implementation” (Pharmaceutical Engineering®, November–December 2018),2 which received Pharmaceutical Engineering®’s Roger F. Sherwood Article of the Year award in 2019.
There is a well-established business case for continuous biomanufacturing, and it has significant support from the US FDA because it is seen as a very effective way to ensure product supply and mitigate drug shortages; more recently, its benefits for shoring up operations close to drug product demand with significantly shorter and more resilient supply chains have also been emphasized.3 A recent report4 indicates that the demand for continuous bioprocessing is increasing, and expenditures in both upstream and downstream continuous bioprocessing equipment are among the top three new expenditures by the companies surveyed. In this article, we focus on the drivers for moving to a continuous biomanufacturing platform and on providing updates in a more compact view about technical aspects. Our aim is to provide insights into life-cycle and regulatory considerations and the potential for continuous manufacturing in emergency preparedness and rapid response efforts.
Design and Operation Considerations
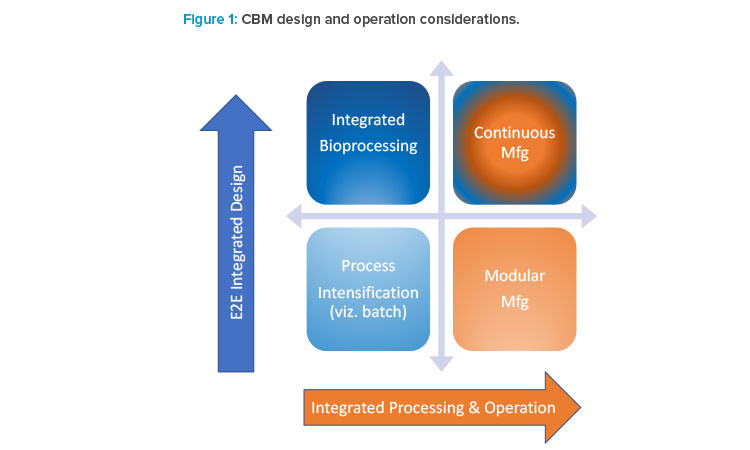
Continuous manufacturing represents the highest level of integrated design and processing currently available in biomanufacturing (Figure 1). The different modalities that map out the boundaries of possible upstream process (USP) and downstream process (DSP) designs vary in terms of end-to-end (E2E) integration and the seamless operability integration of upstream process and downstream process components. Several in-between modalities are defined, especially if upstream process or downstream process is treated separately. Figure 1 defines continuous biomanufacturing in terms of other designs and clarifies the distinction to modular or integrated designs.
Drivers for Moving to a CBM Platform
Continuous manufacturing of biologic products has a number of promising “paradigm shifts” that make the prospect attractive to many organizations. However, sources of resistance can also be identified. Table 1 summarizes some key factors to consider when evaluating whether continuous biomanufacturing is a feasible platform for your specific application.
Table 2 provides one real-world example of the benefits of moving to a continuous manufacturing platform. Improvements in facility area reduction, increase in upstream productivity, downstream column size reduction, and reduced buffer usage are just some of the benefits that can be realized when continuous biomanufacturing is implemented.
| Positive Drivers | Reasons Why Change Is Resisted |
|---|---|
|
|
| Fed-Batch Manufacturing |
CBM |
|---|---|
| Batch cycling time: 15 days Titer: 100% Output per bioreactor: 1× |
Perfusion duration: 60 days Titer (1.5 RV/day): 33% Output per bioreactor: 30× (60 days × 1.5RV × 33%) Productivity increase: 7.5× (FB 4× in 60 days) |
| Titer: 3 g/L | Titer: 1 g/L |
| Process yield: 70% | Process yield: 70% |
| Runs per bioreactor: 18 | Runs per bioreactor: 5 |
| Capacity requirement: 4 × 2000 L SUB | Capacity requirement: 2 × 500 L SUB |
Abbreviations: RV, reactor volume; FB, fed batch; SUB, single-use bioreactor.
Implementing New Technologies
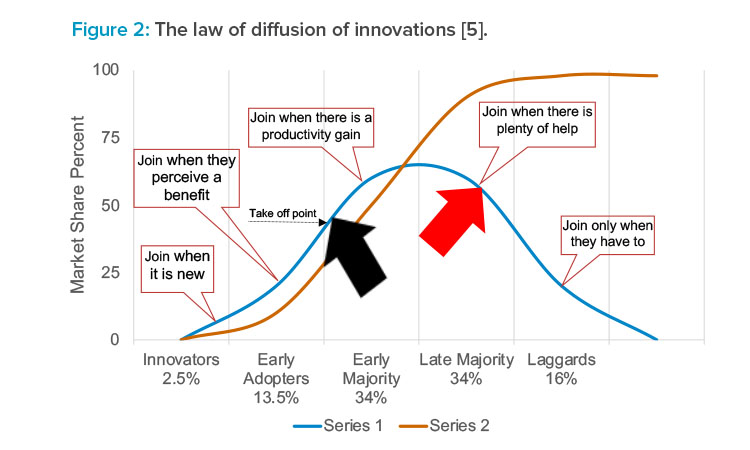
How organizations choose to implement a new technology is based on their business, operational, risk, and market strategies. Generally, all industries fall into one of the five categories shown on the x-axis of Figure 2.5 A company’s decision to implement a continuous biomanufacturing process will be somewhat influenced by which category it belongs to. The black arrow represents where the current movement to continuous manufacturing seems to be for both pharmaceutical and biological products. The red arrow represents where disruptive technologies, single-use systems, and digital sensors, are perceived to be in today’s manufacturing environment. Although a specific organization may not be totally aligned with these advances, the point of implementation can be seen (Table 3).
If continuous manufacturing is implemented, the impact on any new facility assets should also be factored into the decision-making process. When continuous manufacturing requires smaller equipment or less equipment, the facility footprint may be reduced, which can result in facility cost savings. The business case will also discuss the scale of operations and whether the process can be “right sized” in critical unit operations, such as chromatography, to produce the desired outputs while controlling aspects such as column sizing, resin selection, and resin utilization. Resin storage and inventory can be reduced, which further improves the business case. A change of this magnitude will disrupt significantly the current cadence of day-to-day manufacturing. This cadence has a number of elements that must be addressed. Upstream process and downstream process unit operations require advanced scheduling and robust production planning to ensure long-term operational integrity and allow leaner start-up and shutdown sequences in terms of volumetric productivity, product quality, and contamination safeguards.
| Area of Focus |
Key Questions |
|---|---|
| A Is CBM technically feasible? |
When deciding whether to implement CBM, organizations must first ask:
|
| B Is the business case acceptable? |
The next questions should focus on the business case for CBM:
|
| C Is the product risk acceptable? |
Is the risk to the product an acceptable one given the organization’s business and operational strategy? Product knowledge around critical quality attributes (CQAs), critical process parameters (CPPs), and critical material attributes (CMAs) is a must. If moving to a CBM platform impacts product CQAs, the decision should be a no-go. Cell viability must be well understood, going back to the development effort. If the process is not going to be a 100% single-use process, what cleaning impacts will need to be addressed? |
| D Are the process risks acceptable? |
Are the process risks resulting from implementation acceptable?
|
| E Is the process control strategy acceptable |
Does the organization have the ability to develop the robust control strategy needed? |
| F Is the implementation strategy acceptable? |
Life-cycle management considerations: Is the organization able to address these?
|
| G Is the logistic control strategy acceptable? |
Operations management and scheduling need to be considered in CBM.
|
Many manufacturing processes are initially developed in a batch-driven mode of operations and subsequently transferred to a continuous biomanufacturing platform. When implementing a new manufacturing strategy, there must be a plan that includes key elements that address questions that will be asked both internally and during the external regulatory review.
A key driver of continuous manufacturing implementation is the opportunity to reduce the cost of goods. Continuous biomanufacturing allows organizations to make more products faster with lower capital costs and less operator intervention. For example, a company could make more material by using N-1 bioreactors in perfusion mode rather than N bioreactors in batch mode. A new capital facility could be less expensive to build if only an N-1 perfusion mode bioreactor configuration were installed. Continuous manufacturing also has positive impacts in reducing the risk of contamination over successive campaigns because downstream process resin lifetime could be consumed during a single campaign, allowing for timing of new campaigns and resin replacements.
Table 3 includes a series of questions to answer when considering continuous biomanufacturing implementation. If the answers to all the questions posed by activities A through G are positive responses, the decision to implement a continuous manufacturing platform should be seriously investigated and strong consideration given to its implementation.
CBM Platform and Regulatory Requirements
Current regulations and guidelines are supportive of innovative biopharmaceutical development and manufacturing approaches. Although continuous manufacturing is not specifically addressed in guidelines, it fits well into the “enhanced approach.” Some of the following points that are intertwined and integrated into continuous manufacturing may improve regulatory compliance:
- High purity cell lines
- Chemically defined media
- Optimized and stable buffers
- Universal, standardized platform (various proteins)
- Steady state (metabolism)
- Closed system (minimized microbial issues)
- No scale-up (same scale from pilot to commercial manufacturing—scale-out)
- Compatible with disposable technology
- Minimized hold time (eliminate hold tanks and nonessential equipment—improved stability for labile products)
- Continuous flow (minimized residence time)
- High volumetric productivity
- Integrated, modular, simplified operation
- Flexible capacity increase/decrease (within the operating range and then scale-out, if desired)
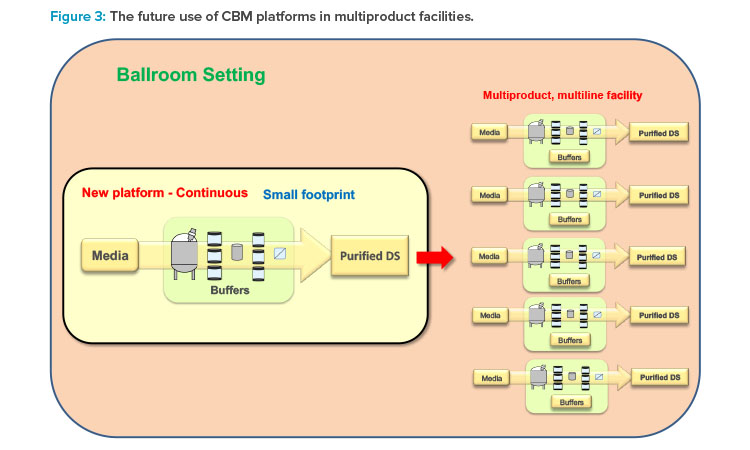
Continuous biomanufacturing platforms have a small footprint (as illustrated in Figure 3) with feedback/feed-forward control that allows for collecting data on the operation/manufacturing process continuously. 2,6 The importance of process analytical technology (PAT) as the main enabler of a robust monitoring and control strategy for continuous manufacturing has been delineated in recent publications.2,7 PAT can support the implementation of continuous manufacturing throughout the entire life cycle. This will provide a state of control of the manufacturing process at all times,2 with benefits including:
- Productivity (cell growth and apoptosis, and all CMAs and CPPs)
- Quality (CQAs)
- Flexibility
- Cost savings (due to reduced equipment and equipment size, footprint, operational services)
- Simplicity
- Mobility (operate at any site near patients)
- Standardization (a technology platform, minimized design and validation)
When capacity demand increases, manufacturing requirements will be accomplished via scale-out (i.e., by repeating the same line). Building an additional production line with the same specifications allows for a quicker design and build, and it simplifies the commissioning, qualification, and validation activities. One can utilize existing protocols and know-how (from baseline) to document and execute all needed requirements. The regulatory approvals process may go faster because no drastic changes have been made.
ICH has initiated the development of a new guidance (ICH Q13) on the topic of continuous manufacturing of drug substances and drug products. Currently projected to reach adoption as a final guideline in November 2022, ICH Q13 guidance will:
- capture key technical and regulatory considerations that promote harmonization, including certain cGMP elements specific to continuous manufacturing;
- allow drug manufacturers to employ flexible approaches to develop, implement, or integrate continuous manufacturing for the manufacture—drug substances and drug products—of small molecules and therapeutic proteins for new and existing products; and
- provide guidance to industry and regulatory agencies regarding regulatory expectations on the development, implementation, and assessment of continuous manufacturing technologies used in the manufacture of drug substances and drug products.
| Regulatory Considerations | |
|---|---|
| Batcha |
|
| Raw materialb |
|
| Process descriptionc |
|
| Control strategyd |
|
| Equipmente |
|
| ICH Guidance |
|
a Definition of batch size should be stated prior to manufacture as a specific quantity of material produced in a process or series of processes, so that it is expected to be homogeneous within specified limits. In the case of continuous production, a batch may correspond to a defined fraction of the production. The batch size can be defined either by a fixed quantity or by the amount produced in a fixed time interval.
b Raw material is a general term used to denote starting materials, reagents, and solvents intended for use in the production of intermediates or drug substance-drug product.
c Refer to “EudraLex Volume 2B: Notice to Applicants and Regulatory Guidelines for Medicinal Products for Human Use. Presentation and Format of the Dossier Common Technical Document (CTD); Differences to a Batch Process” [10].
dManufacturing process produces the product of intended quality in a reproducible way (batch process).
eDiscussed during a preapproval inspection (GMP).
The ICH Q13 position paper8 states:
There is a general consensus that continuous manufacturing (CM) has potential for improving the efficiency, agility, and flexibility of drug substance and drug product manufacturing. Regulatory agencies have seen more companies engaged in the development and implementation of CM in recent years than in the past. Although current regulatory frameworks allow for commercialization of products using CM technology, a lack of regulatory guidelines can make implementation, regulatory approval, and lifecycle management challenging, particularly for products intended for commercialization internationally. An ICH guideline would facilitate international harmonization and could reduce barriers to the adoption of CM technology.
Table 49, 10,11,12 captures from a regulatory perspective the future opportunities for impacting the scientific understanding of the use of continuous manufacturing technologies for the production of biologic molecules.
- A number of issues in the regulatory domain require resolution to make continuous biomanufacturing a viable technology platform for the manufacture of large molecules. These include:
- Differences from batch manufacture: Many continuous biomanufacturing-related definitions and terminologies require further clarification and explanation in the regulatory context.
- Definitions of continuous manufacturing concepts: Examples of key concepts include start-up/shutdown, state of control, process qualification and validation, and continued process verification.
- Harmonizing regulatory common understanding and consistent usage of terminology across different regions: This will lead to improved communication among stakeholders.
- Establishing key scientific approaches for continuous manufacturing: Fundamental scientific approaches for continuous manufacturing may differ from those encountered in batch processes (e.g., concepts of system dynamics, monitoring frequency, detection and removal of nonconforming material, material traceability, process models, and advanced process controls).
- A common understanding of scientific approaches: This will facilitate consistent science, risk-based implementation, and regulatory assessment of continuous manufacturing across different regions.
- Identifying regulatory expectations related to continuous manufacturing: Harmonized regulatory expectations for approval and aspects of life-cycle management that are pertinent to continuous manufacturing can facilitate the adoption of continuous manufacturing and result in consistent regulatory assessment and oversight.
Given the technology and manufacturing of drug substances and drug products for therapeutic proteins, new and existing products need to be addressed. The regulatory expectations with respect to marketing applications and postapproval changes, site implementation, and biopharmaceutical quality systems must also be addressed.
Conclusion
Continuous manufacturing represents the highest level of integrated design and processing currently available. These qualities enable unique functionality and platforms that are potentially capable of rapid deployment and of delivering agile and accelerated timelines from development to on-demand manufacturing. The drivers for adoption of continuous biomanufacturing relate to improved productivity, reduced plant footprint, and overall capital and operational expenditures, many of which are favorable life-cycle considerations. All of these come with more sophistication (i.e., supervisory controls) to enable consistent quality and in-process continued verification. Regulatory considerations for biologics have a higher level of complexity than for small molecules. Therefore, companies considering CBM platforms must be ready to support their applications with robust control strategies with sound evidence-based considerations as well as risk-based justifications.
Though continuous biomanufacturing is not suited for all companies and may only be advantageous to specific products in a portfolio, the future seems quite promising for those organizations committing to enable faster response times to global emergencies and to improved drug product availability (i.e., supply reliability and patient access). As demonstrated in the 2020 ISPE Continuous Manufacturing Virtual Workshop,1 continuous manufacturing and continuous biomanufacturing will contribute to shaping pharma and biopharma as true bioindustry 4.0 technologies, given the level of science, technology, automation, and knowledge management involved and required for their effective deployment. It will be fascinating to witness what the future holds in this regard!
About the Authors






