Improving Access for Patients with Unmet Medical Needs – Overview and Best Practices for Success

This concept paper provides an overview of these programs, the regulatory environment governing them and best practices for design and implementation.
1 The Need for Access
Point to any country on a map of the world. That’s where you might find a patient in urgent need of a new therapeutic option. For many patients, access to new medicines via the well-known, well-defined clinical trial process or through commercial channels is not an option. Consider these scenarios:
- The patient does not live in close proximity to a clinical trial site.
- The patient does not meet trial eligibility requirements.
- The patient was enrolled in a trial, the trial has ended, but availability of commercial supply is delayed.
- The patient lives in a country where a new drug is not or will never become commercially available.
For patients with an unmet medical need who require a new therapeutic option, the wait for approval and commercialization may simply be too long.
Fortunately, mechanisms exist in many countries around the world that enable patients in these situations to gain access to medicines that are in clinical development, are unlicensed in a specific market but licensed elsewhere or are in the Marketing Authorization Application (MAA) process, in a business and regulatory compliant and ethical manner.
Such mechanisms are referred to by a number of terms including compassionate use, named patient use, expanded access and early access, to name a few (Table 1.1). Mechanisms can be put in place for groups (i.e., cohorts) of patients or for individuals (i.e., named patients).
This concept paper provides an overview of these programs, the regulatory environment governing them and best practices for design and implementation.
| Country | Name |
| United States | Expanded Access |
| European Union | Several terms are used, including Compassionate Use, Cohort ATU (France) and Named Patient Supply |
| Japan | Named Patient Access and Personal Importation |
| Australia | Special Access Scheme |
| Canada | Access Program |
It should be noted that these types of mechanisms, often collectively referred to as access programs, are distinct from the off-label use of a medication. “Off-label” describes the use of an authorized drug for an unauthorized purpose, while the programs described here enable access to a drug that is not yet authorized or available in the country in which it is required.
Table 1.2 provides a summary of the differences among compassionate use, off-label use and clinical trials in the EU 1.
| “Compassionate Use” European Regulation | Off-Label Use | Randomized Clinical Trial | |
| Purpose | Serves needs of patients where no alternative treatment exists; available for products applicable for MA via centralized procedure | Serves the needs of patients with an indication other than that which the product is marketed for | Serves the needs of society and future patients and may benefit some of the included participants |
| Party Involved | Patients | Patients | Participants |
| Disease | A life-threatening, chronic, or seriously debilitating disease | Any indication for which the product is not authorized | Any |
| Informed Consent | Required in some member states | Not required | Required |
| License | Medicinal product is not yet licensed | Medicinal product is licensed for other indication(s) | Medicinal product can be licensed and not licensed |
| Responsible Party | Prescribing physician with approval from the regulatory authorities | Prescribing physician | Sponsor with approval from regulatory authorities |
| Control Group | Without control group | Without control group | With control group |
| Data | In some member states, some data are reported to the regulatory authorities | Spontaneous adverse events may be reported | Outcome measure and adverse event data are reported to the regulatory authorities |
| Access to the Intervention | Medicinal product accessed through the program, afterward those patients can have access before the product is licensed | Medicinal product available on prescription | Declaration of Helsinki stipulates that “participants are entitled to…share any benefits that result from the trial, for example, access to interventions…” |
Access programs afford companies the opportunity to meet ethical obligations by providing patients who have an unmet medical need the opportunity to receive a potentially life-saving, life-enhancing or life-extending treatment, in advance of the commercial availability of the product. In addition, such programs can be highly effective in helping foster positive relationships with prescribing physicians and treatment centers, as well as providing an opportunity to gather limited, yet valuable, information about the use of a drug in a wider population.
Demand for access outside conventional clinical and commercial routes may come from patients and physicians anywhere in the world. The trends toward greater transparency of drug development pipelines and broad accessibility of powerful social media tools have led to a more informed, empowered and vocal population of patients. Patients can easily access information about drugs in development via the Internet and are leveraging social media tools such as YouTube, Twitter, and blogs to appeal to companies from which they are seeking access to their products and to call greater attention to their needs. Awareness of new therapeutic options in the pipeline is also facilitated by patient advocacy groups and online forums.
Rather than waiting for inbound requests from patients and healthcare providers, many companies proactively establish access programs as they anticipate demand for their drugs. Among the circumstances likely to stimulate demand are:
- Promising, well-publicized results from clinical trials
- Drugs offering a novel mechanism of action
- Drugs for serious diseases or conditions for which there are no or limited therapeutic options
- Conclusion of a successful clinical trial and the desire to maintain access to the drug for trial participants
- The gap between the New Drug or Biologic Application (NDA/BLA) submission and regulatory approval
- Commercial availability in other territories or regions
- Availability of a new therapeutic option for a rare disease with a patient population spread thinly around the world
Access programs can provide a robust and ethical, local or global solution in all of these situations. Before embarking on development of a program, an understanding of the regulatory environment and review of best practices is an important first step.
2 The Regulatory Environment
Regulations governing access programs are typically set by local competent authorities and define the parameters by which access can be established and how programs must operate. Unlike regulations governing the clinical trial process, which are quite similar across geographies, regulations related to unlicensed medicines can vary considerably from country to country. Below, we offer a sampling of regulations from around the world.
2.1 United States
The US Food and Drug Administration (FDA) categorizes these programs as “expanded access” and describes them as follows 2:
“Use of an investigational drug outside of a clinical trial to treat a patient with a serious or immediately life- threatening disease or condition who has no comparable or satisfactory alternative treatment options.”
The intent of an expanded access program is treatment, in contrast with an investigational drug in a clinical trial, where the primary intent is research.
Since the 1970s, the FDA has facilitated access, under specific circumstances, to drugs or biologics that are still in development. There was no official regulatory recognition of expanded access, however, until 1987 when the
Investigational New Drug (IND) regulations were revised to provide access for a broad patient population under a treatment IND/protocol 3. This revision provided recognition of treatment use for individuals 4, but no criteria or requirements were described.
The 1997 FDA Modernization Act amended section 561 of the FDC Act5 and made expanded access part of the statute, rather than just regulations. An individual patient may obtain an investigational drug for treatment use when:
- The patient’s physician determines that the patient has no comparable or satisfactory alternative therapy.
- The FDA determines there is sufficient evidence of safety and effectiveness to support use of the investigational
- drug.
- The FDA determines that providing the investigational drug will not interfere with the initiation, conduct or
- completion of clinical investigations to support marketing approval.
- The sponsor or clinical investigator submits information sufficient to satisfy the IND requirements.
At the time, the statute only addressed large groups of patients via treatment INDs and did not define the level of evidence required for expanded access.
These rules were clarified by the FDA in 2009. New types of access for treatment use were added to ensure “broad and equitable access to investigational drugs for treatment” 6. The new rule, effective October 2009, provided a uniform standard of evidence for all drug classes and sought to improve access through a better understanding of what was accessible and how.
The new regulations describe:
- Three categories of access (individual, intermediate size and treatment IND/protocol)
- General criteria applicable to all categories and additional criteria that must be met for each
- Requirements for submission
- Applicable safeguards including informed consent, Investigational Review Board (IRB) roles and reporting requirements
A single-patient IND is a request from a physician to the FDA that an individual patient be allowed access to an investigational drug on an emergency use basis. The phrase “compassionate use” is sometimes heard in place of single-patient IND, although officially, “compassionate use” is not a category recognized by the FDA.
When the FDA receives a significant number of requests (~10 to 100) for individual access to an investigational drug for the same use, they may ask the trial sponsor to consolidate these requests, creating an intermediate-size group. If the population of patients to be treated is expected to be less than 100 patients, this is the proper classification to use for this expanded access submission.
When the population of patients to be included in the expanded access use exceeds approximately 100, a treatment protocol or treatment IND is required. A treatment protocol can be submitted when the sponsor seeking access has an existing IND in effect, under which the product is being developed. The request for expanded access and a treatment protocol can be submitted to the already open IND.
A treatment IND is usually submitted to the FDA when the sponsor of the treatment use does not have an existing IND for the drug. In that situation, the sponsor either submits a new IND or requests permission from the drug developer to reference its IND or NDA for any content required by the FDA for regulatory assessment.
Since 2009 when the updated regulations were put in place, there have been between 900 and 1,000 patients treated by expanded access programs each year 7. The total includes single patient INDs, intermediate size and treatment protocols/INDs.
Table 2.1 provides a summary of requirements for Expanded Access Programs (EAPs).
| Requirements for all US EAPs 8 | ||
| ||
| Requirements for Individual Patient EAPs 9 | Requirements for Intermediate-Size Populations 10 | Requirements for Treatment IND 11 |
|
|
|
Companies can charge for drugs provided under expanded access, recovering direct costs of making the drug available. For intermediate-size and larger programs, the sponsor also can recover monitoring and IND-related costs. The amount to be charged must be justified to the FDA and approved. In addition, the sponsor must provide reasonable assurance that charging will not interfere with developing the drug for marketing approval. In the case of treatment INDs and protocols, there must be assurance that the development process is progressing, including evidence of enrollment in clinical trials.
FDA views expanded access programs as a “community responsibility” that encompasses the patient, his or her doctor, the sponsor, FDA and the IRB 12.
| Community Member | Their Perspectives and Roles |
| The Patient |
|
| The Doctor |
|
| The Sponsor |
|
| FDA |
|
| Institutional Review Board |
|
2.2 Europe
In 2001, Directive 2001/83/EC was introduced, stipulating that “A Member State may, in accordance with legislation in force and to fulfil special needs, exclude from the provisions of this Directive, medicinal products supplied in response to a bona fide unsolicited order, formulated in accordance with the specifications of an authorized health care professional and for use by his individual patients on his direct personal responsibility.”
This provision effectively enabled a healthcare professional to supply an unlicensed medicine on a named patient basis. Subsequent updates to the legislation included further provision for the distribution and supply of unauthorized products in response to the spread of pathogenic agents, toxins, chemicals and radiation that may cause harm. In 2004, Article 83 of Regulation 726/2004 introduced the legal framework for the provision of “compassionate use” in the European Union, for any medicinal products that are eligible to be authorized via the Centralized Procedure.
The European Medicines Agency (EMA) defines compassionate use as 13:
The use of an unauthorized medicine outside a clinical study in individual patients under strictly controlled conditions. Compassionate use programs are for patients in the European Union (EU) who have a disease with no satisfactory authorized therapies or cannot enter a clinical trial. They are intended to facilitate the availability to patients of new treatment options under development.
According to the EMA guidance on compassionate use, the objectives of Article 83 are to:
- Facilitate and improve the access of patients in the European Union to compassionate use programs
- Favor a common approach regarding the conditions of use, the conditions for distribution and the patients targeted for the compassionate use of unauthorized new medicinal products
- Increase transparency among member states in terms of treatment availability
Although products authorized via regulatory routes other than the centralized procedure are not specifically covered by Regulation 726/2004, for applicable products, the regulation provides a framework for the centralized assessment of a promising new drug, to enable widespread access across Europe.
The implementation of compassionate use, however, remains the responsibility of each individual member state. Any alternative routes to accessing an unlicensed medicine are governed by the legislation in the member state where the patient and treating physician reside.
Within Europe, there are typically two major routes to accessing an unlicensed medicine. The first is named patient supply, where a treating physician will usually request access to a medicine on an individual patient basis. The other route is typically designed to capture groups or cohorts of patients rather than individuals, and permission to supply often requires a more in-depth assessment from the relevant regulatory agency.
One thing is certain: the differences in the legislation and processes for supplying an unlicensed medicine in Europe are diverse and complex.
European Compassionate Use Legislation does not include any binding processes or procedures to guide individual countries on how to implement mechanisms that enable access to unlicensed medicines. Instead, each country decides individually whether to allow this type of access and how the programs should be structured. The result is tremendous diversity and variability in the processes of each country, introducing the need to consider the requirements and demands for a promising new drug on a country-by-country basis.
In 2010, the European Clinical Research Infrastructures Network (ECRIN) published results of a survey on regulatory requirements of compassionate use programs [11]. It was clear that different countries have adopted different requirements and that “compassionate use” is not interpreted in the same way across Europe. The authors concluded that “there are more differences than similarities in ‘compassionate use’ programs in Europe.”
Other than differences in terminology and the legal routes for supply, access schemes across Europe differ in many aspects. For example, in some countries, the drug must be provided at zero cost, while other countries allow a company to charge for access to the product. Where charging is possible, this needs to be carefully considered as, dependent on the country in question, the price may impact future discussions on pricing and reimbursement, or be referenced by other countries.
The timing and requirement for pharmacovigilance and safety reporting also varies by country, with some countries requiring quarterly reports on the use of the drug. Similarly, labeling requirements vary. For named patient supply, it is often possible to supply a product in its original packaging, but for some of the cohort programs, specific labelling must be used and in some cases, be agency approved.
While many decisions are made at the member state level, there are a number of guiding principles which are echoed in the centralized legislation, and which apply in general terms to each country. These include, but are not limited to, the following:
- The drug for which access is being sought must have shown promise, with clinical trials either completed or ongoing, or the product should be the subject of an application for a marketing authorization. For EMA compassionate use, the product should be in phase III. Only in exceptional circumstances would a drug in phase II be considered.
- The patient requiring treatment must have a serious, life-threatening or life-changing disease or condition.
- There should be no alternative licensed treatments available or open clinical trials through which the patient could receive appropriate treatment.
It is clear that access programs can positively impact patients and provide a critically important solution for their treating physicians. It is also clear that demand for earlier access to promising medicines is increasing. In some cases, the figures are significant. For example, the former French Health Products Safety Agency (now the National Security Agency of Medicines and Health Products, ANSM) reported that by 2007, more than 20,000 patients had been treated with over 200 products under compassionate use legislation introduced in 1994 14.
Other countries around the world also have schemes and processes in place which enable the supply of unlicensed medicines to their citizens. Some of these programs, including those in Canada and Singapore, are very well-established, while others are still under development.
3 Progress and Change
Legislation and guidance governing the regulatory routes for early access is constantly evolving, and ranges from refinement of the existing processes to the introduction of new routes for supply where previously none existed.
In recent months, changes have been seen in the guidance and/or processes for unlicensed supply in Brazil, Germany and Peru and further changes are anticipated in Japan, China, Eastern Europe and the Middle East.
As recently as April 2014, the MHRA launched a new Early Access to Medicines (EAMS) scheme in the UK, which introduced a two-part process to facilitate access to unlicensed medication.
The first part is the introduction of a Promising Innovation Medicine (PIM) designation, which can be assigned to a product following the assessment of clinical data and which can provide an early indication that the specific product has potential for the EAMS scheme.
The second part is where a scientific opinion is issued following the assessment of the benefit: risk profile of the medicine. This process aims to make positive scientific opinions available on the MHRA’s website, to assist doctors and patients in making treatment decisions and informing them of the risks and benefits of the product.
Given the myriad of rules and regulations surrounding early access and their nuanced interpretation, it is important for a company considering an access program to have a thorough understanding of what is allowed in each country in which an access program will operate and fully understand the associated obligations. For countries with well-established programs, such as the US, Germany and France, the rules are clear; however, in those geographies where program parameters are not firmly defined or are evolving, setting up a program can be challenging and simply identifying the established legislation is not enough, as guidelines can be open to interpretation. Companies should also remain informed of new efforts to facilitate access such as the “right to try” movement gaining interest in some states in the US.
4 Best Practices
Supply of medicines under an access program can deliver significant benefits to patients in need. In order to ensure the successful development and implementation of a program, ensure patient safety and minimize risk, a number of factors must be considered. Figure 4.1 reflects the range of considerations that must be taken into account to help ensure a successful access program.
Figure 4.1: A Number of Considerations Must Be Taken Into Account When Designing an Access Program
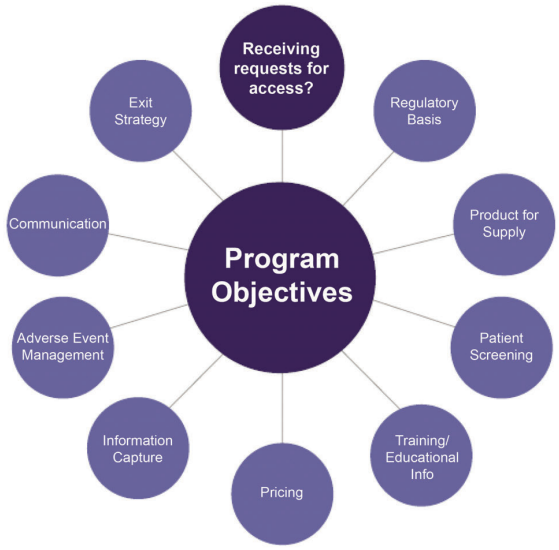
4.1 Engage Internal Stakeholders
Successful access programs require the cooperation of several functional areas, including manufacturing, medical affairs, clinical, regulatory, quality, commercial teams and supply chain. Including a representative from reimbursement or pricing is important if the company charges for the drug, as this help ensure the pricing is done in a way that is consistent with future pricing plans.
A cross-functional team is necessary to put in place safety, education, data collection and reporting and submission requirements, define clinical criteria for patient eligibility, determine the geographic reach of the program and ensure enrollment in any ongoing clinical trials will not be compromised. Once the geographic scope of the program is identified, communication and coordination with local affiliates will help ensure a smooth roll-out and ongoing program success.
If possible, an internal person should be assigned to manage the program, being responsible for timelines and communication among stakeholders. Seamless coordination of key functions, along with timely reporting of program metrics back to stakeholders, will help ensure success. Unfortunately such a resource may not exist and when this is the case, or when the company lacks experience in developing and managing such programs, an external partner can add significant benefit and expertise, and reduce the exposure to risk.
4.2 Define the Scope and Timing
At the outset of an access program, it is essential to clearly define, agree upon and communicate the scope and aim of the program within the organization. As the program progresses, more internal groups (e.g., affiliates) will become aware of it and may want to influence its direction, change the scope or discuss aspects of the program with their local regulatory bodies. As patients and patient organizations learn about the program, demand is likely to grow and may come from a wide range of geographies. In parallel with a growing demand, patients who may not fit the program’s eligibility criteria might seek access. All of these situations must be considered and planned for when structuring the program.
While programs can evolve and expand over time to better meet patient needs, it is important to remain mindful of the agreed upon aim, to ensure consistency and fairness in delivery of the program. As the program progresses, a regular review of milestones, successes and areas for improvement should be conducted.
Planning for an access program should begin early. If the need is likely, decisions about the program should be made at the beginning of pivotal trials. This allows time for preparation of standard operating procedures, consultation with regulatory authorities (where required) for approval of the program and development of information for healthcare providers regarding dosing, administration and restrictions.
When developing a program, initial discussions with internal stakeholders should focus on how the company wants to allow access and should include the following:
- Geographies in which the program will operate
- Patient eligibility requirements
- Pharmacovigilance and safety reporting requirements and processes
- Processes by which physicians will request access and the documentation they will need
- Training that will be needed for local affiliates, medical directors and others
- Timing and exit strategy for completion of the program
The set-up phase is also the time to identify decision makers, routes of communication and what the escalation process will be, should an unexpected event take place. This phase is a very intense period of time for the team involved and may take six to nine months of investment.
In addition to planning the launch and management of the access programs, companies should proactively determine when and how the program will end. In many cases, programs end upon receipt of marketing authorization or when reimbursement has been finalized. The impact that the end of the program will have on individual patients receiving treatment should be considered and patients should clearly understand how their participation will conclude.
4.3 Communicate with External Stakeholders
Promotion of an investigational drug and unlicensed supply is not allowed by regulatory authorities. As such, companies offering an access program cannot actively solicit physician or patient interest. However, companies can provide information on their websites and at medical conferences in response to requests. US-based programs can be listed on clinicialtrials.gov and some other agencies list the approval of such programs on their own websites.
Companies have issued press releases about their access programs; however, careful attention to wording is essential to ensure the content is educational and not promotional. Claims about safety and efficacy are not allowed.
As a new drug is advancing in the pipeline or is awaiting approval, it is likely patient advocacy groups will be monitoring its progress. Companies also may be in active communication with these groups during the clinical trial process. As a result of this ongoing engagement, advocacy groups are natural partners to help educate both physicians and patients on these access programs, manage expectations and be a trusted resource for providing information.
4.4 Manage Supply
The availability of product to support the access program should be an early consideration, especially for those products where commercial supply is not yet established, such as those in development or without marketing authorization in any territory. In such a situation, a product may be in short supply and lead times can be lengthy, so early involvement of supply chain/CMC groups is essential.
Physician-initiated requests for access can come at any time from any geography. Forecasting and collaboration with clinical teams and local affiliates will help define expected demand and ensure adequate supply. In contrast, demand is more predictable when the access program is offered by the company to a cohort of patients or to those who had participated in a clinical trial and are now awaiting commercial availability.
A timeline of expected country-by-country transition to the commercial supply also should be maintained to more effectively manage supply.
5 Program Logistics
For pharmaceutical and biotechnology companies accustomed to shipping bulk quantities of drugs to a wholesaler or a limited number of clinical sites, a shipment earmarked for an individual patient presents a unique challenge. From a logistical standpoint, a number of factors are critical to success when managing the logistics of a global access program.
5.1 Security
In a mass market situation with bulk shipments, the supply chain is fairly predictable; consolidated shipments routinely go to the same wholesalers and to major population centers. In many cases, shipments are made at ambient temperature and are not under urgent time pressures.
With global access programs, shipments are made for individual patients (or on a patient-by-patient basis) via their physicians and local pharmacies; as a result, the supply chain becomes longer and typically requires more steps. In many cases, the patient is in dire need of the drug and so the supply chain must operate at a much more rapid pace. The drug must be managed efficiently through customs and cannot be subjected to temperatures that may be damaging.
When managing a program, it is important to maintain complete visibility of all steps in the supply chain along with storage conditions, from the moment the drug leaves the warehouse to the time it gets to the patient’s physician.
5.2 Timing
Patients who are part of an access program may require very rapid delivery of the drug, in some cases, within 24 to 48 hours. With a longer supply chain and patients often located in distant time zones, efficiency is critical.
Anyone who has shipped even the most simple of packages overseas knows that the customs process can be a bottleneck. In some countries, customs are more unpredictable with individually interpreted import controls. In these cases, it is important to closely monitor the situation to prevent the shipment from getting delayed. Relationships with local freight carriers and distribution agents are essential for timely delivery as they can facilitate documentation procedures.
A successful program must leverage a dynamic, flexible approach to logistics, integrating both global and local or regional capability. Operating at the level of individual packs/shipments for individual patients, these programs start with a global network of freight carriers, but must become localized at the far end of the supply chain. Once shipments have arrived into one country, individual orders go onward toward different hospitals and may need to be handled at different temperatures.
5.3 An End-to-End View
In order to strengthen the supply chain, a best practice is to put in place an agreed method of onward transport with distribution partners. Checks should be put in place for the conditions to be fulfilled before partners are allowed to take product from distributors. It is also crucial to carefully monitor the pathway a drug takes to get to the service provider’s warehouse from the manufacturer. A product shouldn’t be accepted into the supply chain unless it is known exactly where it is from and it is possible to prove where it has been from the moment it left the manufacturer.
It is also necessary to have a thorough understanding of the import regulations, as defined by the countries into which the drug will be shipped. In some cases, it may be necessary to ship direct from the country of origin, which needs to be factored into any logistics plan.
When managing logistics for an access program, the goal must be to minimize risk and get the drug to the patient on time. The clock is ticking from the moment the request comes in and at the end of the supply chain is a patient in need. Supply chains should be “challenged” on a regular basis to reveal any weak points, identifying critical hand-off points and which transactions may present difficulty. In conducting these exercises, one can then assign resources where needed most, to ensure a smooth delivery and a successful outcome.
6 Conclusion
For patients with life-threatening illnesses, license approval or commercial launch of an innovative new drug may come too late. The patient may not meet the eligibility criteria for a clinical trial or live in a geography where no formal launch is planned. In the EU, a drug may not clear reimbursement hurdles in an individual country until well after its approval is granted.
In all of these scenarios, global access programs provide a route to obtain innovative drugs prior to their approval or launch, potentially helping patients who have run out of therapeutic options.
During development of an access program, a number of key parameters must be established and clearly defined, including the scope of the program, patient eligibility, timing and logistics. Combine these success factors with a thorough understanding of regulatory requirements and open, proactive communications with internal stakeholders, patients, physicians, advocacy groups and regulatory authorities, and the access program will be poised to deliver significant benefits.
Limitation of Liability
In no event shall ISPE or any of its affiliates, or the officers, directors, employees, members, or agents of each of them, or the authors, be liable for any damages of any kind, including without limitation any special, incidental, indirect, or consequential damages, whether or not advised of the possibility of such damages, and on any theory of liability whatsoever, arising out of or in connection with the use of this information.
© Copyright ISPE 2014. All rights reserved.
No part of this document may be reproduced or copied in any form or by any means – graphic, electronic, or mechanical, including photocopying, taping, or information storage and retrieval systems – without written permission of ISPE.
All trademarks used are acknowledged.
Acknowledgements
By the ISPE IP Community of Practice
This concept paper was written and reviewed by members of the ISPE Investigational Products (IP) Community of Practice (COP).
Elizabeth Cooper Clovis United Kingdom
Mark Corbett Clinigen Group United Kingdom
Marie DeGayner Kuker Kuker Regulatory Consulting USA
Delphine Fabry Sanofi France
Rachel Huskisson Clinigen Group United Kingdom
Sheri Smith Courante Oncology USA


