Stage 2 Process Validation: Process Performance Qualification Batches

The purpose of this paper is to stimulate further discussion and suggest potential practical application. Approaches to providing an answer are proposed, but more experience in implementation of the lifecycle approach to PV is needed to reach a consensus position.
This discussion paper proposes ideas for answering the question “How many process performance qualification batches (PV stage 2) are needed to demonstrate a high degree of assurance in the manufacturing process and that the control strategy is sufficiently robust to support commercial release of the product?” Considerable input has already been received, considered, and/or incorporated. The authors are interested in hearing about other approaches that could be used, and lessons from use of the proposed approaches described in the discussion paper. The paper may be modified or expanded sometime in the future to reflect additional input.
Additional input in the following areas is sought:
- Examples of how the approaches described in this paper have been used or modified, especially for:
- Other types of DP processes
- API processes
- Biopharma processes
- Revalidation of legacy processes
- Other approaches, statistical or non-statistical, that may be used to justify the number of PPQ batches
- Using data from stage 1 batches to justify fewer PPQ batches
- Other science, risk, and/or statistical criteria for between-batch variability to help provide rationale for the number of PPQ batches
Please direct all feedback to pvstage2@ispe.org. The authors would prefer that all input be focused primarily on the themes identified above.
Note: Version 1 of this document has been replaced to correct a statistical calculation error.
1 Introduction
Since the adoption of the ICH Q9, Quality Risk Management (QRM), by the Pharma industry, the importance of the QRM approach and its benefits has become evident. This trend invites re-examination of well-established practices. One such example is the widely adopted concept that validation is a one- time activity and that three consecutive successful validation batches is sufficient to demonstrate process reproducibility. It has long been recognized that successful manufacture of three consecutive batches may not necessarily provide assurance of process reproducibility, as routinely relying on three sequential batches alone does not always provide strong confidence that the process will continue to deliver product that consistently meets quality acceptance criteria.
The revised Process Validation (PV) Guidance from FDA (January 2011) aligns process validation activities with a product lifecycle concept, emphasizing the expectation that process validation starts with process design and spans the entire lifecycle of the marketed product. More specifically, the Guidance recommends that Process Performance Qualification (PPQ) approaches (PPQ being an activity that is part of what the Guidance describes as Stage 2, where process design is evaluated to determine if it is capable of reproducible commercial manufacture) should be based on well-grounded scientific justification, an appropriate level of product and process understanding and adequate demonstration of control. The FDA Guidance does not define a regulatory expectation for the number of process qualification batches. It is expected that manufacturers make a rational decision for the number of validation batches and design of the PPQ study based on product knowledge and process understanding. A sufficient number of batches should be included in the study(ies) to demonstrate reproducibility and an accurate measure of between batch variability. This will provide sufficient evidence that the process is reproducible and that commercial release of the product to the market is justified.
1.1 Scope
The science and risk based approach described in this paper is applicable to the manufacture of human and animal drug and biological products, including drug products, the drug constituent of a combination (drug and medical device) product, active pharmaceutical ingredients (APIs) and drug substances. It is applicable both to validation of new manufacturing processes and to validation of changes to existing processes (e.g. changes in site, process scale, equipment, etc.).
The suggestions described in this paper focus on how the task of justifying a number of PPQ batches might be addressed and are not intended to represent an industry consensus. The approaches described in this paper are intended primarily for prospective validation. For concurrent validation other approaches not described here may be more relevant.
The application of these recommendations assumes that a manufacturer has established a robust Quality Management System as described in ICH Q10 for documentation, training, etc.
1.2 Background
The 2011 FDA PV guide advises us to look at knowledge acquired from development and historical performance of a process to help define the expectations for process validation. This information is assessed in the context of the product’s clinical use (or from other sources of product knowledge) and its potential impact on patient safety and product efficacy. For example:
- “Process knowledge and understanding is the basis for establishing an approach to process control…. Strategies for process control can be designed to reduce input variation (or) adjust for input variation during manufacturing… Process controls address variability to assure quality of the product.”
- “Manufacturers of legacy products can take advantage of the knowledge gained from the original process development and qualification work as well as manufacturing experience….”
- “…activities …. such as experiments or demonstrations at laboratory or pilot scale also assist in evaluation… and prediction of performance of the commercial process.”
During the process qualification stage, the process design is evaluated to determine if the process is capable of reproducible commercial manufacturing. The goal of PPQ is to confirm the process design and demonstrate that the commercial manufacturing process performs as expected. This includes providing scientific evidence that the process is reproducible and will consistently deliver quality products. How much process knowledge/understanding and other evidence is needed to achieve this aim? For the purpose of this paper, this question can be restated as:
- How many qualification batches are required for the PPQ studies, when considered along with Stage 1 activities, to demonstrate that the process implementation and control strategies are sufficiently robust?
Based on the extent of process knowledge and process understanding, there may be cases where the number of validation batches needed to show process reproducibility may be less than or greater than three. Any applicable regulatory requirements for a minimum number of validation batches should also be taken into account.
1.3 Overview
This discussion guide is intended to provide suggestions that will stimulate further thought and discussion of this topic; it does not reflect a consensus position of the industry. This paper describes a framework for systematically assessing the level of product knowledge and process understanding, and how well the control strategies are linked to the Critical Quality Attributes (CQAs). The residual risk identified from this assessment may then be translated to a number of validation batches. The overall approach described in this paper is outlined in Figure 1. Following this, data from the PPQ batches are analyzed using appropriate statistical methods to determine the out-come of the PPQ study and to help identify what the appropriate level of sampling and analyses may be in Stage 3 (the commercial manufacturing stage of the product lifecycle). A discussion guide “Applying Continued Process Verification Expectations to New and Legacy Processes” that describes some practical approaches to fulfilling the requirements during Stage 3 was issued at the same time as original issue of this discussion guide. For a process where different steps may be validated separately, such as can be the case for a multiple-step drug substance process, the evaluation may indicate that different steps necessitate different numbers of PPQ batches, based on the science and risk associated with each step.
Figure 1: Workflow for Determination of the Number of Stage 2 PPQ Batches
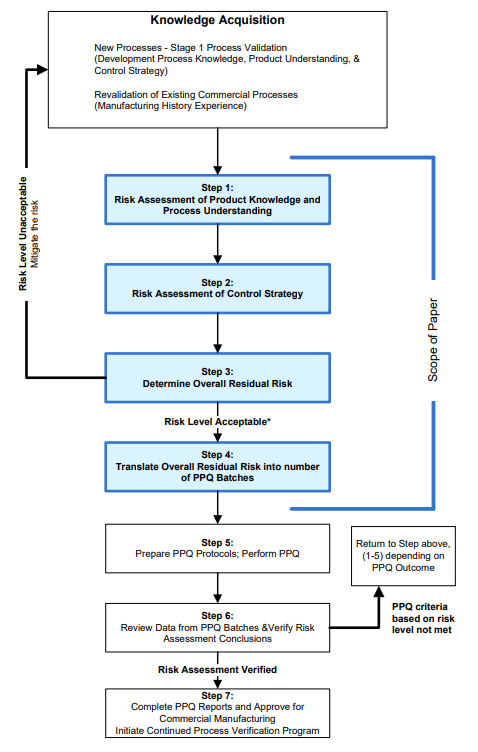
* Determination of an acceptable level of risk may be based on internal company standards. The standards may be designed to encourage additional development work (increasing product and process understanding) rather than performing large numbers of PPQ batches.
2 Risk-based Approach
It is suggested that the risk assessment described here be performed periodically during development in order to highlight the extent of understanding and how it might impact the PPQ program. If high risk(s) is/are identified from the assessment, it may be prudent to increase knowledge before starting the stage 2 PPQ activities, in order to reduce the risk and subsequently the number of PPQ batches required to demonstrate process reproducibility.
Risk assessment can be performed in different ways. For this discussion, it is assumed that the assessment will focus on three inter-related considerations: product knowledge, process understanding, and process control strategy. An example of the risk ranking system is included in Appendix 1 with exemplification of the characteristics of the ranking assignment for the following aspects:
- Step 1 – Assess Product and Process Knowledge and Understanding Risks
- Step 2 – Assess Control Strategy Risk
2.1 Assess Product Knowledge (Step 1a)
Identified product quality requirements as outlined in ICH Q8 (R2) are referred to as the Quality Target Product Profile (QTPP) and can be further related to the Critical Quality Attributes (CQAs) of the drug product or drug substance. At a minimum, severity and probability (that variation might impact product safety, efficacy or quality) of an identified risk should be included in the risk assessment. The degree to which acceptable ranges for CQAs are based on experimental data (lab, clinical, non-clinical), predictions/information from proven models, and other sources of knowledge is related to the risk that the ranges are appropriately justified. Even in circumstances where the drug’s mechanism of action is not well understood, efforts made to understand CQAs may help to mitigate the risk of affecting safety or efficacy.
2.2 Assess Process Understanding (Step 1b)
Process understanding attempts to establish the relationships between material attributes, process parameters and CQAs and to estimate their variability without application of the control strategy. This can be used to estimate one portion of risk. However, it is admittedly difficult to estimate process variability in the absence of the control strategy, but separating these different considerations is one way to ensure that the different factors are all considered. The user may choose to group factors (considerations included in the risk assessment) together differently or pursue a different tact for evaluating similar considerations.
Process understanding can be established from several sources, including those listed here and explained further in Appendix I:
- Development phase – understanding of variability from development and product characterization, primarily from stage 1 of the product lifecycle (the product/process development stage). An important component of this is having sufficient analytical capability to permit monitoring of the consequences of variability. Process understanding from the development phase may be lacking for legacy products or for generic manufacturers, which puts increased importance on gaining process understanding from other sources.
- Prior Knowledge – understanding from prior experience, such as with other similar processes or from use of platform technology. For a mature product, data from annual product review, product quality review, deviation investigation, complaint investigation, and/or change control information may be used as sources of process understanding.
- Degree of process understanding /unit operation – extent of knowledge gained/explored during the development of each unit operation and the depth of understanding of the effects of inputs and process parameters on process results. Impact on variability from personnel (e.g. from equipment set-up, monitoring of process and/or product handling), from selection of appropriate equipment, from the performance of that equipment and from other factors (e.g. environmental considerations such as influence of humidity on process performance) are considerations to be included.
- Process predictability and modeling – the sophistication of the laboratory model/small scale model and its ability to adequately predict the effects of input variability on outputs at commercial scale.
- Effect of scale changes – understanding the effect of changes to the scale on which the process is run.
2.3 Assess Control Strategy Risk (Step 2)
The Process Control Strategy evolves through the development of process and product knowledge in Stage 1 of the product lifecycle. Its main purpose is to control the impact of input variability from materials, environment, and operational practice so that the output variability of the product attributes and process performance is appropriately monitored and controlled. It encompasses all aspects of manufacturing – from raw material specifications through finished product release, including parameter controls for all unit operations in the process. Factors in the control strategy and their potential impact on product and process variability are especially important considerations in determining the appropriate number of Stage 2 PPQ batches.
Factors that could be considered for risk assessment of the control strategy include:
- Raw material specification
- The impact of variability of critical material attributes, how well this variability is managed, and the potential impact that raw material attributes may have on the process and on product quality can be a significant contributor to control strategy risk.
- Equipment capability vs. process requirements
- Prior to commercialization, manufacturing processes are introduced through technology transfers that might include engineering, qualification, and/or demonstration batches produced at either full scale or at a scale that is considered representative of full scale. Manufacturing facilities and process equipment are qualified for the commercial process. Equipment capability derived from these qualifications can be compared with process requirements as one contributor to risk determinations.
- Experiences with process performance
- Experiences with the process in managing variability, with appropriate control of scale effects, and comparable process performance serve as indicators that the control strategy established for the process should be sufficient for reproducible process performance. Consistent success during preparation of development batches (or with historical performance for a legacy process) (i.e., no history of recurring or unexplained problems and incidents have been satisfactorily addressed) implies there is reduced risk for demonstrating process reproducibility with validation batches.
This is only intended as an example of factors that may be considered in identifying risks related to process reproducibility. A company may choose different considerations than described here in performing their risk assessment, and/or may choose to put considerably more, or exclusive, emphasis on the control strategy, rather than weighting controls strategy equally with the other contributions (product and process knowledge and understanding) proposed here.
2.4 Determine Residual Risk Level (Step 3)
Following the risk assessment of the factors of the product/process and control strategy understanding, a residual risk level should be determined. This residual risk level reflects the confidence in performance of the commercial process and can be used to determine the appropriate number of PPQ batches. With higher residual risk that is not minimized or mitigated, it is reasonable to expect more batches will be required to confirm that the process is capable of reproducible commercial manufacture.
Choosing only to increase the number of PPQ batches is not a substitute for insufficient process development or understanding. Reasonable efforts should be made to identify and mitigate identified higher risks before attempting process qualification.
Application of a Quality by Design (QbD) approach to development of the process, product, and control strategy should result in a lower residual risk by improving process understanding and therefore minimize the number of required PPQ batches.
The output of the risk assessment – residual risk levels – should be determined from FMEA or another Quality Risk Management tool in alignment with QRM principles.
An example of considerations that may contribute to this risk assessment is provided in Appendix 1. For this discussion, five overall residual risk levels are identified as shown in Table 1:
| Residual Risk Level | Description |
|---|---|
| Severe (5) | Multiple factors have high risk ratings. |
| High (4) | Few factors have high risk ratings or all have medium risk rating. |
| Moderate (3) | Medium risk level for multiple factors or high risk level for one factor |
| Low (2) | Medium risk level for a few factors, the others are low risk |
| Minimal (1) | Low risk level for all factors |
A residual risk level will represent the level of remaining risk revealed from the assessment of product knowledge, process understanding and control strategy effectiveness. The assessment should:
- Consider all product CQAs, either in combination or evaluating each CQA individually. If assessed individually, the assessment should include the CQA with the highest associated residual risk, and the number of validation batches should be commensurate with the highest residual risk CQA.
- Be performed by a team that includes subject matter experts from areas such as manufacturing, technical services, process development, quality assurance, statistics, and process validation.
- Provide justification for the evaluation and ranking of each factor.
Identification of a severe or high residual risk indicates there are significant gaps in knowledge and/or control strategy. Additional effort should be invested in improving knowledge and/or the control strategy before preparing PPQ batches.
3 Approaches to Determining Number of Validation Batches (Step 4)
The basis for claiming that the control of between-batch variability is appropriate for commercialization of a manufacturing process requires comparison of values of CQAs from a series of replicate batches. With the understanding that a meaningful value for the between-batch variability of all the CQAs will not be known until Stage 3 of the Process Validation lifecycle (when enough data points have been collected to permit appropriate statistical analysis), sponsors may choose different approaches for establishing a “high degree of assurance” that the process will meet this criterion. Below, we describe three different approaches for establishing whether the level of between-batch variability is appropriate for commercialization based on a small number of PPQ batches. Each approach has assumptions regarding how a high degree of assurance is established. In selecting an approach for determining the number of batches required, organizations need to accept the underlying assumptions of the approach or else either modify the approach (such as by changing assumptions) or choose an alternate approach.
Following the risk assessment of the product understanding, process knowledge and control strategy, the overall residual risk is identified and should serve as a basis for determining the number of PPQ batches. A process that has higher residual risk may require more PPQ batches in order to provide enough assurance that the between batch variability is appropriately controlled before commencing the
commercial distribution. On the other hand, a process that has low residual risk after stage 1 would only require a few PPQ batches to confirm the effectiveness of the control strategy before the decision for commercialization can be made.
The connection of the number of PPQ batches and the assurance they provide may be addressed with a rationale describing why the number of validation batches chosen is appropriate, and/or with statistical considerations. Examples of these approaches are described here, including two statistically-based options. Other approaches may also be appropriate.
In most cases, it may be appropriate to include some data from development batches along with data collected from the PPQ batches to improve the statistical power of the data and thus reduce the number of batches for the PPQ. Justification of inclusion of development batch data as a means of reducing the number of PPQ batches should be based on an assessment of the differences between those batches and the PPQ batch study design (including data collection and sample analysis).
Other circumstances that are outside of the scope of this discussion may also influence the decision for the number of PPQ batches needed. These considerations include different dosage strengths, different package sizes, different batch sizes, use of different but similar equipment, etc. It may be appropriate to use approaches such as bracketing or “matrixing” in these circumstances. The statistical acceptance criteria chosen for evaluating between batch variability may also influence the number of batches identified for the PPQ study.
It is difficult to define a generalized acceptance criterion that shows process reproducibility in a statistically meaningful manner. For CQAs where reproducibility can be shown or calculated from data collected over a reasonable number of batches, acceptance criteria may be included for the PPQ. But not every CQA will be amenable to statistical treatment with a small number of batches; for these, demonstrating reproducibility may be extended to Stage 3 of the product lifecycle.
The extensive sampling and testing of PPQ batches designed to meet statistically based criteria (such as for criteria established for examining within-batch variability) will also provide data to support the acceptable performance of the process. Consider the number of samples needed for meeting these statistical acceptance criteria, as this may also influence determination of the number of PPQ batches.
For the statistical treatments described here, it is assumed that results fall within a normal distribution and parameters being examined are centered to target. Alternative statistical methods would be needed if these assumptions are not valid.
3.1 Approach #1 – Based on rationales and experience
This approach is based on the assumption that, for low risk processes, the preparation of three consecutive PPQ batches is appropriate. In other words, successfully preparing three batches can provide an acceptable degree of assurance to show reproducibility for low-risk processes, as has been shown by the historic success of using this number of batches for many validation studies. In these
situations, the PPQ exercise becomes one assessing the results from the PPQ batches against expectations, to verify that the control strategy for a well-understood process is appropriate for ensuring product quality. It is also important to recognize that regulatory authorities in some regions expect that for most situations, a minimum of three PPQ batches will be prepared to show process reproducibility.
Using a rationale-based approach, one can construct an argument based on historical success with low risk processes and acknowledgement that increased residual risk should be accompanied by an increased number of PPQ batches, e.g. shown in Table 2:
| Residual Risk Level | Number of Batches | Rationale |
|---|---|---|
| Severe (5) | Not ready for PPQ | Additional development should be pursued to identify processes or controls needed to reduce residual risk. |
| High (4) | 10 | Higher residual risk makes it unlikely that a small number of PPQ batches are adequate to show process consistency. A larger number of successful batches may show process consistency, but achieving this would be unlikely if controls are not adequate. A preferable course of action would be to perform additional development and/or knowledge acquisition to reduce residual risk so that fewer PPQ batches would be needed. |
| Moderate (3) | 5 | Increased residual risk can be addressed by preparing two additional PPQ batches to provide further demonstration of process consistency. |
| Low (2) | 3 | Knowledge and control strategy are regarded as sufficient. Three PPQ batches have been shown historically to be appropriate for demonstrating process consistency for many low-risk processes. |
| Minimal (1) | 1-2 | Strong knowledge and high degree of controls minimize risk. One situation where this may be appropriate is for verifying specific controls associated with a well-understood change to a process, or where process can rely on using a control strategy successfully shown for a similar product or process. Processes with PAT as a significant part of the control strategy may also be of minimal risk. |
An example based on the PQLI Illustrative Example (PQLI IE) is provided in Appendix 3 to explain how risk assessment of product and process knowledge and control strategy as described in the PQLI IE, can be applied to determine number of PPQ batches.
3.2 Approach #2 – Target Process Confidence and Target Process Capability
The FDA guidance states that “Before any batch from the process is commercially distributed for use by consumers, a manufacturer should have gained a high degree of assurance in the performance of the manufacturing process such that it will consistently produce APIs and drug products meeting those attributes relating to identity, strength, quality, purity, and potency”. This statement poses two questions:
- What is an objective measure that the process will consistently produce product that meets its requirements?
- What is an acceptable high degree of assurance?
These two questions are termed as target process performance (question 1) and target process confidence (question 2).
3.2.1 Target Process Performance
Statistically, one measure of process robustness that can be used to assess the ability or capability of the process to meet the required quality requirements is Process Capability (Cpk).
By definition a capable process has a Cpk of 1.0 or greater. Therefore demonstration of a Cpk of 1.0 as a starting point for assessing the capability of a process undergoing validation seems reasonable. However, the level of confidence in this assessment should be commensurate with the risk associated with the level of knowledge, understanding and robustness of the control strategy as discussed earlier. Where the risk of process failure has been established as low, an estimated process capability of Cpk ≥ 1.0 with 90% confidence does not seem to be unreasonable based on the limited experience and data available at commercial product launch. However, as the residual process risk increases so should our need to provide a higher level of confidence that the process is actually performing at an acceptable level of capability.
The level of residual process risk may be correlated to a residual risk analysis such as that as suggested as described above and in the example in Appendix 1.
3.2.2 Target Process Confidence
The other factor to consider is the level of confidence needed in the Cpk calculated using the Stage 1 and Stage 2 data.
At what point do we need to reach the high level of confidence of quality consistency between batches? Is it at completion of Stage 2 PPQ, or during Stage 3 Continued Process Verification? Certainly each individual batch manufactured during Stage 2 is expected to meet the quality requirements before being released for commercial distribution. However, determining how robust the process actually is may take considerable time and a number of batches to experience the full range of variability inputs into the
process and the resultant impact on the product CQAs. In fact one could argue that the range of input variability that the process will experience is infinite and therefore can never be completely known. Therefore, some middle ground must be sought that will provide high confidence in individual stage 2 PPQ batches and reasonable confidence in the process such that the validation batches can be released for commercial distribution. The reality is that we can only build very high levels of confidence with time and experience. Therefore if we can accept the premise that depending on the product and process risk it may be acceptable to begin commercial distribution prior to reaching an extremely high confidence level such as 99%, then what is the trigger that allows us to say that Stage 2 PPQ activities been successfully completed?
While any confidence level selected is somewhat arbitrary, it seems reasonable that at least a 90% confidence in the capability of the process to meet the quality standards considers both patient risk and process robustness. At the same time, within-batch process capability data and enhanced sampling required for the stage 2 PPQ batches will help to ensure appropriate quality to support commercial product release. This approach provides additional confidence in the quality of individual validation batches and support for reasonable confidence in the robustness of the process to continue to produce batches meeting quality requirements.
Table 3 provides an example of how target confidence levels can be determined based on the risk assessment. Processes with Minimal residual risk (i.e. all risk categories evaluated at the lowest level), should not require additional assurance during PPQ beyond a demonstration that commercial systems and procedures are appropriate, so statistical justification of the number of batches is not required.
| Residual Risk Level | Target Confidence | Comments |
|---|---|---|
| Severe (5) | N/A | A Severe or High risk ranking indicates major gaps in knowledge and understanding. These are cases where additional effort on product/ process/control strategy development may be necessary. A high target confidence level is needed to provide a high degree of assurance that a higher risk process will perform reproducibly, and help to assure consistent product supply. |
| High (4) | 97% | |
| Moderate (3) | 95% | Target confidence levels here are designed to provide reasonable assurance of the process capability, supporting commercial distribution; higher assurance of process capability would be achieved eventually with more Stage 3 commercial batches. |
| Low (2) | 90% | |
| Minimal (1) | N/A | A Minimal risk ranking indicates high confidence of existing understanding and the capability of the control strategy. It is not necessary to base the number of PPQ batches on a target variability. |
3.2.3 Determine Number of PPQ Batches
A statistical method based on confidence intervals for Cpk (see Appendix 2 for a detailed discussion) provides a justifiable number of batches utilizing the Readily Pass criteria, the Target Process Performance, and the Target Process Confidence. Assuming a Readily Pass criteria of greater than or equal to 1.6, and Target Process Confidence from Table 3, Table 4 below illustrates the minimum number of batches required to assure that we are confident that Readily Passing processes are Capable (Target Process Performance of Cpk ≥ 1.0). The number of required batches will change if different criteria are used for the Readily Pass criteria, the Target Process Performance, or the Target Process Confidence. It should be noted that the minimum number of batches stated below can be achieved through a combination of Stage 1 and Stage 2 activities.
| Residual Risk Level | Minimum Number of Batches | Target Process Confidence for Cpk 1.0 | Acceptance Criteria | ||
| Readily Pass Calculated Cpk | Marginally Pass Calculated Cpk | Fail Calculated Cpk | |||
| Severe (5) | Not ready for PPQ | NA | |||
| High (4) | 14 | 97 | ≥ 1.6 | 1.6 > Cpk ≥ 1.0 | < 1.0 |
| Moderate (3) | 11 | 95 | |||
| Low (2) | 7 | 90 | |||
| Minimal (1) | 1-3 | NA | |||
Note: See Appendix 2 for details on how the table was derived. Tables 3 and 4 provide just one example of linking the target Confidence and Cpk level with the residual risk ranking. It is possible that other Confidence and Cpk levels are also appropriate, as long as sufficient justification is provided.
The following is an example of the application of Table 4. If a product and process has been determined to be of moderate residual risk at the end of stage 1, and if the data from at least 11 batches calculates to a Cpk of at least 1.6, then there is at least 95% confidence that the true Cpk is at least 1.0. However, if the calculated Cpk is greater than or equal to 1.0 but less than 1.6 there is still potential that the process has acceptable capability, but the data provides less than 95% confidence that the true Cpk is at least 1.0. Finally, if the Cpk is less than 1.0, then there is less than 50% confidence that the true Cpk is at least 1.0. In this case process improvement should be considered before proceeding.
The criteria in Table 4 may be interpreted as follows:
- If the PPQ batches meet the “Readily Pass” criteria, the process is qualified and has been adequately shown to be reproducible, then sampling and testing may be adjusted to a statistically appropriate level for Stage 3 (routine release). An enhanced Stage 3 sampling and testing plan may not be needed, provided that the impact of all input variability has already been thoroughly verified.
- If the PPQ batches meet the “Marginally Pass” criteria, continue full qualification level sampling and testing on those attributes not meeting “Readily Pass” until significant variability estimates are achieved. (Assumes the range of input variable values is within the range characterized as acceptable for the process.)
- For any process not meeting “Marginally Pass” the process has demonstrated a lack of capability. The process would necessarily need to be improved before continuing validation.
- No target Cpk or acceptance criterion is proposed for a minimal residual risk level. The minimal risk ranking indicates high confidence of existing understanding and the capability of the control strategy, and in such a case the PPQ exercise becomes one of verifying the control strategy, with high confidence of success.
- Note: There are two fundamental underlying assumptions used in this approach. First the process is assumed to be in-control and second the data is normally distributed.
3.3 Approach #3 – Expected Coverage
Another statistical approach that could be used is based on the concept of order statistics (ref 1). When a set of n observations from an unbounded population are ranked in ascending order, x1, …., xn, the expected probability of an observation being less than or equal to xm is m/(n+1), where m is the ranking order. Consequently, the expected probability of a future observation being within the range defined by [x1, xn] is (n-1)/(n+1). This expected probability is independent of the underlying distribution (for instance, a normal distribution of data is not required). This expected probability can be considered as the “coverage” of the population that the range of the existing data can provide.
It is intuitive to understand that with increasing number of PPQ batches, the probability of a future batch with results within the PPQ experiences increases as well. This increasing probability provides increasing “coverage” (or assurance of the future batch behavior) as well as increasing assurance of the overall process robustness. Therefore, a direct connection between the number of PPQ batches and the desired assurance can be made. For example, a low residual risk process does not require high degree of assurance from the PPQ batches alone, while a process with high risk after stage 1 lifecycle would demand high assurance from the PPQ batches. While there is some arbitrariness to any choices of the expected coverage, a high expected coverage (say 95% or higher) for a severe risk ranking process seems to be reasonable, while a 50% expected coverage for a low risk ranking process provides some degree of assurance. Firms are encouraged to determine the appropriate level of expected coverage that is meaningful to their own processes. Table 5 provides an example of determining the number of PPQ batches based on the risk assessment, where expected coverage is not given to the severe risk
category to be consistent with the previous two approaches. For example, a passing high-risk process would have 50% confidence that 4 of 5 values (80%), are within release limits.
| Residual Risk Level | Expected Coverage from PPQ Batches Alone, with 50% confidence | Number of PPQ Batches |
|---|---|---|
| Severe (5) | N/A | Not ready for PPQ |
| High (4) | 80% | 9 |
| Moderate (3) | 70% | 6 |
| Low (2) | 50% | 3 |
| Minimal (1) | N/A | 1-3 |
Note: The number of batches in table 5 is the number of PPQ batches only.
Once the desired number of PPQ batches is prepared, it is prudent to evaluate the data and determine if there is any unexpected between-batch variability exhibited by the PPQ batches. One straightforward and effective way is to plot the data, such as box plots, multivariate charts and other plots that would provide visual summary of the within and between batch variations. Additionally, several statistical options exist for evaluating the between batch variability. Although the order statistics used to determine the number of batches is a distribution-free statistical method, it is appropriate that the PPQ results be analyzed using statistical methods that may rely on a specific distribution, to maximize the information from the data. For example, when normal distribution is confirmed, the Cpk discussion from approach #2 can be used. Another example is the Narrow Limit Gauging (ref2), or pre-control (ref3). As an example, the narrow limit can be set as the middle 50% of the specification range, for a CQA with two-sided specification. Table 6 provides the readily pass and marginally pass requirements, assuming a normal distribution.
| Number of Batches | Readily Pass Number of batches allowed to be outside of middle 50% spec range | Marginally Pass Number of batches allowed to be outside of middle 50% spec range |
|---|---|---|
| 9 | 0 | 3 |
| 6 | 0 | 2 |
| 3 | 0 | 1 |
- The Readily Pass requirement is established that a normally distributed process with a failure rate of 0.27% or lower would have at least a 50% chance to pass the requirement.
- The Marginally Pass requirement is established that a normally distributed process with a failure rate of 1.64% or lower would have at least a 90% chance to pass the requirement.
- If the PPQ batches do not meet the Marginally Pass requirement, further assessment is required, and the process may require improvements.
- It is expected that specifications are met for all PPQ batches in all cases. If any specification is not met, the Narrow Limit Gauging concept described in Table 6 is not applicable.
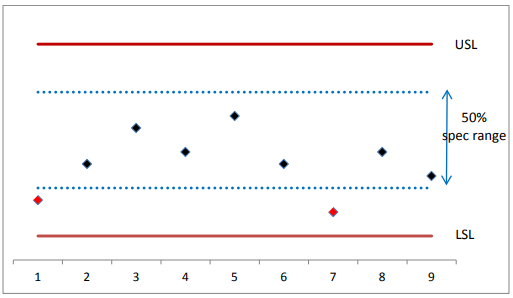
The above concept is illustrated in Figure 2. Nine PPQ batches were manufactured, and all passed the specification requirement for the CQA. Two out of the 9 PPQ batches were within the specification range but outside of the middle 50% of the specification range, while the rest were within the 50% of the specification range. Therefore, the PPQ batches marginally passed the requirement, and enhanced monitoring of this CQA is recommended for the stage 3 of PV lifecycle.
Figure 2. Illustration of Readily Pass and Marginally Pass Requirements
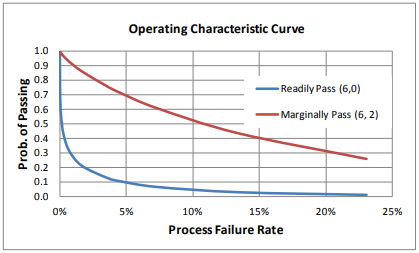
The relationship between the process capability and the probability of such a process meeting the readily pass and marginally pass requirement can be demonstrated graphically. Assuming normality, Figure 3 illustrates the probability of meeting the readily pass and marginally pass criteria when 6 PPQ batches are executed. Similar curves can be developed for other readily pass and pass requirements, as well as for other distributions. Firms are encouraged to develop these curves to justify the requirements chosen for the performance criteria.
Figure 3. Probability of meeting Narrow Limit Gauging criteria, assuming normal distribution
4 Conclusion and further discussion
The approaches described in this discussion guide utilize the concepts of Quality Risk Management and Lifecycle management along with quantitative measures to justify the number of batches included in a PPQ study prior to product commercialization. The level of residual risk associated with the manufacturing process is used to choose a quantifiable degree of assurance that the process will ultimately demonstrate a robust (acceptable) level of output variability. The higher the residual risk, the more statistical assurance required during PPQ, prior to commercialization.
One approach for assessing the level of residual risk is described. While the 2011 FDA PV Guidance does not indicate a statistical approach is needed, two possible statistically-based approaches for determining the number of PPQ batches are discussed. Another experience-based approach that does not use statistical criteria for determining the number of PPQ batches is also included. In practice, the approach chosen to translate the residual risk to a number of PPQ batches and the suitability of this approach for the product and process should be justified, whether it is one of the three described here or another approach should be one that is most suitable for the particular circumstances of the product and process to be qualified.
Companies may identify other approaches for justifying the number of validation batches from consideration of product and process knowledge and understanding risks and the control strategy to be used. They are encouraged to share thoughts with other ISPE members on improving these approaches and identifying alternative approaches (see discussion forum on ISPE website).
5 Appendix 1
The purpose of the risk assessment described here is to provide an example of how characteristics for risk ranking assignments can be defined and used to determine a relative risk ranking.
The relative risk ranking is done systematically as illustrated in Figure 1 and described in Section 2, focusing on:
- Product knowledge (Figure 1, step 1 and Section 2.1)
- Process understanding (Figure 1, step 1 and Section 2.2)
- Control strategies effectiveness (Figure 1, step 2 and Section 2.3)
In each step the knowledge and decisions from PV stage 1 are compared to the ranking characteristics to determine the relative risk ranking for product, process and control strategy respectively.
5.1 Step 1a – Evaluation of product knowledge
The evaluation of product knowledge focuses on severity of harm to the patient and probability that variability has an impact on the safety, efficacy and quality of the product.
The risk ranking level is assigned based on an evaluation of methodology applied to identify CQAs and an evaluation of how well the impact of variability is understood.
The table below provides an example of characteristics used to guide the ranking assignment.
| Product Knowledge Factor | Relative Risk Ranking Characteristics of ranking assignments | ||
| Low Risk | Medium Risk | High Risk | |
| Identification of CQA and impact of CQA variation on patient |
|
|
|
| Product characterization |
|
|
|
5.2 Step 1b - Evaluation of Process Understanding
Development Phase
Much of the process understanding will come from the product and process development and characterization phases of the product validation lifecycle (Stage 1). These activities provide information and knowledge about the potential impact of the expected variability of the input materials and manufacturing process on critical quality attributes of the product as the process is designed to run during commercial manufacturing. Establishing process understanding should include exploration of the relationship between variability of process parameters, material characteristics, and other areas of variability and the potential effect on CQAs.
Predictive models can be developed that simulate variation of inputs to the process and impact to the critical quality attributes of the product. This is especially true for new drug products developed under Quality by Design (QbD) principles.
Development and clinical batches provide estimates of process variability and capability. Criticality levels for process parameters are derived from development, characterization, and manufacturing experience (pilot, demonstration, and clinical trial material batches).
For legacy products, full-scale manufacturing data may be useful in establishing the effects of input variations on product attributes.
Prior Knowledge
In many cases as new products are developed and commercialized, the manufacturing platform chosen for the commercial process is familiar to the firm. The process technology is likely to have been used to manufacture other products and considerable knowledge has been realized. This knowledge base can be drawn on to make estimates of the expected performance of similar processes being developed and validated.
Inherent process variability can be estimated from the prior knowledge, including experiments and knowledge of similar products and processes. This along with consideration for variability the process will experience due to process operators and shifts, process interruptions, hold times, multiple equipment sets, and multiple material suppliers can also help define what number of validation batches should be planned. If the variability from any of the sources is high, or knowledge about the impact or range of the variability is low then this increases the risk of demonstrating process reproducibility.
The risk ranking level is assigned from an evaluation of degree of process understanding, process predictability and understanding of effect of scale as well as effect of variability on process output.
The table below provides an example of characteristics used to guide the ranking assignment.
| Process Understanding Factor | Relative Risk Ranking Characteristics of ranking assignments | ||
| Low Risk | Medium Risk | High Risk | |
| Degree of process understanding / unit operation |
|
|
|
| Process predictability and modeling |
|
|
|
| Process response to input variability |
|
|
|
| Effects of scale changes |
|
|
|
Rankings are aligned with definitions and characterizations provided in ASTM E2475, “Standard Guide for Process Understanding Related to Pharmaceutical Manufacture and Control”. For the purpose of this discussion guide, the characterizations in the ASTM publication have been condensed from 5 to 3 Risk Rankings and some aspects have been included in the “Control Strategy” evaluation rather than here.
5.3 Step 2 - Evaluation of Control Strategy effectiveness
The control strategy should be designed, based on product and process understanding, to ensure that a product of required quality can be produced consistently. The evaluations of control strategy effectiveness therefore focus at suitability of the defined control strategies to manage variability.
The risk ranking level is assigned from an evaluation of methodology applied for setting material specifications and process controls as well as evaluation of equipment capability versus process requirements.
The table below provides an example of characteristics used to guide the ranking assignment. This table is not meant to be a comprehensive listing of all the factors of the control strategy for a manufacturing process. Rather it attempts to identify those factors most likely to contribute to differences in variability between processes.
| Control Strategy Factor | Source of Potential Variability and/or Uncertainty | Relative Risk Ranking Characteristics of ranking assignments | ||
| Low Risk | Medium Risk | High Risk | ||
| Raw Material specifications |
| Specifications of material attributes impacting product quality justified based on development data | Limited justification of specifications of material attributes. |
|
| Equipment Capability vs. Process Requirements | Capability of equipment to control operating parameters within acceptable ranges | Comparison of parameter control ranges from equipment qualification with process requirements indicates all parameters are well within equipment control capabilities and supported by qualification data | Comparison of control ranges from equipment qualification with process requirements indicates marginal capability to meet requirements for a limited number of process parameters | Comparison of parameter control ranges from equipment qualification with process requirements indicates a significant number of parameters are similar to equipment control capabilities |
| Experiences with process performance to date |
|
|
|
|
| Monitoring capability and detect-ability | Ability of monitoring tools and methods to detect variation | Attributes measured in real time at a sensitivity where performance variability is likely to be observed | Attributes measured off- line (after batch completion) at a sensitivity where performance variability is likely to be observed. | Attribute measurement sensitivity and/or accuracy is inadequate to use for controlling performance. |
6 Appendix 2
Bissell4 determines the approximate lower confidence bound for Cpk as:
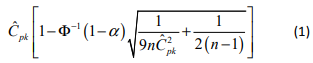
Using these equations we can algorithmically solve for the minimum number of observations during validation, n, that it will take to provide confidence that the process capability exceeds the desired Target Process Performance. Taking values for 𝛼 based on the Target Process Confidence (95% confidence = 0.05 𝛼) and Ĉpk based on the Readily Pass criteria (Cpk ≥ 1.6 implies a Ĉpk of 1.550), equation 1 above can be solved for the smallest n such that the value of equation 1 is greater than or equal to the desired Target Process Performance (≥ 1.0, or a value of 0.950 in the equation).
Note: There are two fundamental underlying assumptions used in this approach. First the process is assumed to be in-control. The second is that the data is normally distributed.
7 Appendix 3
Example: Risk-Based Approach to Determine Number of PPQ batches based on PQLI Illustrative
Example
To demonstrate the risk-based approach for justifying and determining the appropriate number of Stage 2 PPQ batches, a case study based on the PQLI® Product Realization using Quality by Design: Illustrative Example5 is presented here. The Illustrative Example describes a thorough development approach that results in a low-risk process. For detailed information about designed experiments, design spaces, risk assessments done during development including identification of critical process parameters and material attributes, and relevant elements of the proposed control strategy, please refer to reference 5.
The example illustrates the development of a tablet formulation, 30mg (small molecule), called “PaQLInol”, using the enhanced, QbD approach. PaQLInol is a BCS Class 2 compound (low solubility and high permeability). The drug product manufacturing process includes the following steps: dispense, blend, lubricate, compress, and coat.
Although the PQLI Illustrative Example discusses quality attributes systematically, the illustrative example focuses on the highest risk critical quality attributes of Dissolution and uniformity of dosage units (UDU). A risk assessment (Table A1) identifies Dissolution as the highest risk quality attribute, and therefore this example focuses on Dissolution. Table A1 shows the cause and effect risk assessment matrix for PaQLInol tablet 30mg after formulation development and before process development
| Drug Product | ||||||
| CQA/Unit Operations | Dispense | Blend | Lubricate | Compress | Coat | Package |
| Appearance | M | |||||
| Identity | M | M | ||||
| Assay | M | M | M | |||
| Impurities | ||||||
| Uniformity of Dosage Units | H | M | M | |||
| Dissolution | H | H | ||||
| Microbiology | ||||||
The Illustrative Example describes extensive formulation development which leads to an immediate release formulation with Dissolution acceptance criteria of Q=80% at 20 minutes and identification and use of algorithm as an important element of the dissolution control strategy. The discussion here is based on information found in the Illustrative Example.
7.1 Evaluation of Product and Process Understanding
Using the risk assessment matrix given in Appendix 1, Tables A3.1, 3.2 and 3.4 are developed using information given in Part 2, Illustrative Example, further explanation being given below the tables.
| Product Knowledge Factor | Relative Risk Ranking Characteristics of ranking assignments | ||
| Low Risk | Medium Risk | High Risk | |
| Identification of CQA and impact of CQA variation on patient |
|
|
|
| Product characterization |
|
|
|
From an evaluation of impact of CQA variation on patient, the product understanding risk factors are all assigned as Low Risk, based on the following arguments:
- Physiochemical, biological and pharmacokinetic information is used to input to process development using the enhanced, QbD approach.
- The role of excipients in providing an acceptably formulated product is well understood based on designed experiments and the formulation is optimized based on this information
- The impact of variation in dissolution profiles on bioavailability was assessed in an in vivo study. Acceptance criterion for the in vitro Dissolution CQA is established as Q = 80% in 20 minutes from this study. The in vitro dissolution method is able to differentiate between tablet variants (IVIVR variants) with acceptable and unacceptable in vitro release rates and has a relationship to in vivo behavior.
All product understanding factors are considered to be Low risk level, and the Relative Risk Ranking for Product understanding is therefore Low.
A similar evaluation is performed to assign a risk ranking for process understanding.
| Process Understanding Factor | Relative Risk Ranking Characteristics of ranking assignments | ||
| Low Risk | Medium Risk | High Risk | |
| Degree of process understanding / unit operation |
|
|
|
| Process predictability and modeling |
|
|
|
| Process response to input variability |
|
|
|
| Effects of scale changes |
|
PaQLInol Tablets |
|
The process understanding factors are assigned as Medium to Low Risk, based on the following arguments explained in the Illustrative Example:
- Causes of interrelationships between variables are identified and explored sufficiently by designed experiments (DoE) to be expressed in a design space algorithm for dissolution. The algorithm contains factors for:
- Drug substance particle size
- Magnesium stearate specific surface area
- Lubrication time
- Crushing force of tablets
The ranges assessed in the DoE of the above four factors are included in the proposed design space for dissolution as acceptable ranges. The largest contribution to prediction of dissolution values using this algorithm comes from drug substance particle size.
- A design space is proposed.
- PAT tools are applied during process development (Stage 1), but are not relevant for controlling dissolution. Dissolution is highly dependent on 3 material attributes – drug substance particle size, magnesium stearate surface area and tablet crushing force, all of which are process- and scale-independent. Lubrication time is easily determined and controlled.
- Material specific critical quality attributes are identified.
- Effect of scale changes is predictable.
Three process understanding factors are considered to be Medium risk level, one factor is considered
Low. Therefore the Relative Risk Ranking for Process understanding is assigned as Medium.
7.2 Evaluation of Control Strategy
Several control strategy options are considered in the Illustrative Example and 4 are discussed. Option 1, a feed forward mechanism, is preferred. In this option, the API particle size distribution (PSD) value is obtained from its Certificate of Analysis, the MgSt SSA (magnesium stearate specific surface area) value is determined on receipt, and assuming a target crushing force of 85N (Newtons), the lubrication time is calculated based on the algorithm to target 90% dissolution. The dissolution value for release is calculated from the equation using actual lubrication time and crushing force, instead of being measured using a QC method. Tables A3.3 and A3.4 summarize this control strategy. A detailed risk assessment regarding critical process parameters is available in the Illustrative Example.
| CQA | Acceptance Criteria | Control Strategy Element | Control Strategy Acceptance Criteria | Control Type |
|---|---|---|---|---|
| Dissolution | Q = 80% in 20 minutes | API particle size (D0.9) | 5 – 30 μm | CoA value |
| MgSt SSA | 3000 – 12000 cm2/g | Test on receipt | ||
| Lubrication Time | 1 – 8 minutes | Calculated to target 90% dissolution | ||
| Crushing Force | 60 – 110 N | Target 85N |
| Control Strategy Element | Source of Potential Variability and/or Uncertainty | Relative Risk Ranking Characteristics of ranking assignments | ||
| Low Risk | Medium Risk | High Risk | ||
| Raw Material specifications |
|
|
|
|
| Equipment Capability vs. Process Requirements | Capability of equipment to control operating parameters within acceptable ranges |
|
|
|
| Experiences with process performance to date |
|
|
|
|
| Monitoring capability and detectability | Ability of monitoring tools and methods to detect variation | Attributes measured in real time at a sensitivity where performance variability is likely to be observed PaQLInol Tablets | Attributes measured off- line (after batch completion) at a sensitivity where performance variability is likely to be observed. | Attribute measurement sensitivity and/or accuracy is inadequate to use for controlling performance |
Risk ranking for all control strategy factors is assigned as Low Risk from the following arguments:
- • Raw materials: Critical raw materials attributes have been identified and the impact of these critical attributes; PaQLInol particle size distribution and magnesium stearate surface area, on dissolution has been explored extensively in development. This factor is Low risk.
- • Equipment capability vs. process requirements: Equipment capability is not fully described in the PQLI Illustrative Example. However, process understanding indicates that there is low risk of impact of the process on Dissolution CQA when a diffusion mixer, and gravity and power- assisted press are used. Different equipment of these designs may influence the algorithm, which requires verification for any new piece of equipment For the purpose of this example, equipment capability is assumed to meet the requirements of process parameter control, the major process parameter being lubrication time which can be set automatically or be controlled manually, both facile equipment capabilities to achieve. This factor is Low to Medium risk
- • Experience with Process Performance to date: Although not extensively discussed in the illustrative Example the formula and process are used to manufacture batches for phase 3 clinical studies. It is unlikely the final proposed control strategy will be fully applied for clinical manufacture and the scale may not be that proposed for commercialization; however, it is assumed that all batches are manufactured satisfactorily and product complies with CQA acceptance criteria. Scale impact is well-understood. This factor is Low risk
One factor in this risk assessment of the control strategy is considered Low to Medium risk level and the others are Low risk. The Relative Risk Ranking for Control strategy is therefore assigned as Low risk.
7.3 Determine Residual Risk Level for PaQLInol Illustrative Example and Recommendation for Number of validation batches
From the above risk ranking assessments the residual risk level is assigned as Low due to Medium risk level for a few factors and the majority of factors are Low risk. The number of validation batches required for dissolution is defined accordingly:
| Risk Factors | Assigned Risk Level |
|---|---|
| Product understanding | Low |
| Process understanding | Medium |
| Control strategy risk | Low |
| Overall residual risk level (see Table 1) | Low |
With the overall process and product risk ending at Low, the recommended number of validation batches can now be determined using for instance one of the approaches described in this paper.
| Approach | Number of Batches |
|---|---|
| 3 batches (according rationales in Table 2) |
| 7 batches needed to meet a pre-established process capability with Cpk 1.0 at 90% confidence level. (See Tables 3 and 4) Using this approach assumes that data are distributed normally. Total number of batches includes PV stage 1 and PPQ batches which are evaluated as a collective data set. |
| 3 PPQ batches needed to achieve an expected coverage at 50%, which according to Table 5 is sufficient degree assurance from PPQ batches, for a low overall process and product risk. |
Validation of dissolution is assessed by dissolution testing of 60 individual tablets pulled from across each of the stratified sampling points.
Validation dissolution acceptance criteria provides a 90% confidence that there is at least a 95% confidence of meeting USP Dissolution Requirements (see reference 6) of Q = 80% in 20 minutes. The validation acceptance criteria are for n= 60, sample mean ≥ 85% & RSD of mean ≤ 8.5. Dissolution data are plotted graphically to monitor batch-to-batch performance variability.
Samples from each batch should be tested for dissolution and the results compared to the model prediction to confirm that expected results provide verification of the model. These data can be used when determining what enhanced monitoring or routine monitoring will be used in Stage 3 of the product lifecycle.
It is assumed that the facility, utilities and equipment are qualified.
7.4 Conclusion
This case study describes an example with extensive product understanding and formulation development and could be regarded as a model approach for development, so determination of a low number of validation batches is reasonable.
The case study shows how use of a risk assessment tool leads to a reasonable number of batches for Stage 2 PPQ.
If the extent of knowledge indicated in this example is not available for a process, an assessment would likely identify higher risks, a concomitant increase in the overall risk determination for the process and a higher minimum number of validation batches needed to demonstrate process reproducibility. 1, 2, 3 , 4, 5,6
Acknowledgements
Mette Bryder, (Lundbeck)
Harold Etling, (Eli Lilly)
Jeff Fleming, (Pfizer)
Yanhui Hu, (Abbott)
Peter Levy, (PL Consulting)


