Validation & Data Integrity in eClinical Platforms

To address the trial specific setups, eClinical solutions have to be built from various integrated technologies and tools designed to be utilized in clinical trials, working together sharing data, eliminating duplication of activities, and streamlining the use of multiple technologies for the end user.
1 Introduction
It is well understood that each clinical trial is conducted and managed as an independent project even if several trials investigate the same Investigational Medicinal Product (IMP). Each clinical trial is different as each addresses different parts of the development cycle or varying product indications or endpoints. Trial projects, especially across the various phases, vary greatly in terms of duration, number of patients to be recruited, the pace of enrollment, and the spread of geographic location(s) involved.
To address the trial specific setups, eClinical solutions have to be built from various integrated technologies and tools designed to be utilized in clinical trials, working together sharing data, eliminating duplication of activities, and streamlining the use of multiple technologies for the end user (Figure 1.1).
Figure 1.1: Mapping of Sample Sytems to the Clinical Process
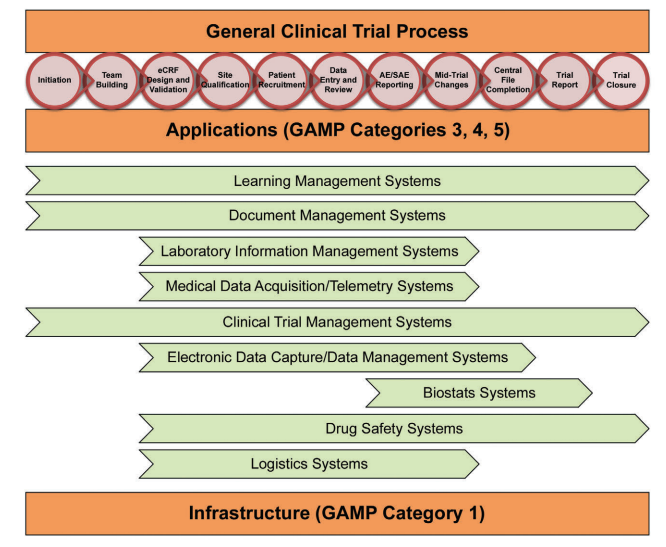
The need to ensure data integrity through the life cycle of a clinical trial and across all the systems involved is of paramount importance as inconsistent, incorrect or corrupted data could endanger the safety of patients, adversely affect the outcome of the trial and increase the risk of a failure during the submission procedure. Therefore, this aspect has increasingly become the focus of regulatory oversight. One of the main drivers for this has been that the industry has embraced individual or strategic outsourcing of clinical trial activities to Contract Research Organizations (CROs) and sponsors as well as CROs also adopting Software as a Service (SaaS) offerings especially in the area of Electronic Data Capture (EDC) or Interactive Voice Response Systems/Interactive Web Response Systems (IVRS/ IWRS). Often this leads to a chain of partners with an increasing risk of losing direct control for the sponsor. Even when strategically partnered with a CRO, the responsibility to address these risks resides with the Sponsor and cannot be delegated. This requires extensive and increasing efforts for oversight, which must be considered when addressing the risks with regard to data integrity.
While the application of the GAMP® 5 principles 1 to the validation of GCP relevant systems has already been discussed in an ISPE Concept Paper, “The Application of GAMP® 5 to the Implementation and Operation of a GxP Compliant Clinical System,” 2 the challenges in the setup and maintenance of an eClinical Platform are largely not addressed.
2 eClinical Platforms
The introduction of the GAMP® Good Practice Guide: IT Infrastructure Control and Compliance, has also seen the term “platform” officially associated with the IT infrastructure of GxP regulated environments for the first time. A platform provides the technological environment (hardware and software) required for an application to fulfill its intended use. The efficient and quality-assured operation of IT infrastructure is facilitated by the use of reusable building blocks, which consist of logical groupings of standardized system components.
The introduction of GAMP® 5 extended the GAMP® software Category 1 to include infrastructure software (infrastructure software tools and layered software), thereby effectively achieving an interface to IT infrastructure. The so called “layered software” includes software such as operating systems, table calculation applications or statistical programming tools, which constitutes platforms for the development of applications.
The main difference between this and an eClinical Platform is the fact that the “layered software” focuses on individual software products (instances) in their condition at delivery, while the eClinical Platform constitutes a preconfigured application portfolio as an intermediate layer between the clinical trial process and IT infrastructure.
For the purpose of this paper, an eClinical Platform is defined as a pre-existing environment of integrated computerized systems that can be adapted to support the conduct of a clinical trial by utilizing existing, validated functionality and processes.
Typical platforms include Electronic Data Capture (EDC) system, Clinical Trial Management System (CTMS), electronic Trial Master File (eTFM), statistical systems as well as safety systems and others. Individual components of the eClinical Platform may require set-up or configuration to meet the requirement of the individual clinical trial.
Figure 2.1: Flow of Information through the Different System Layers
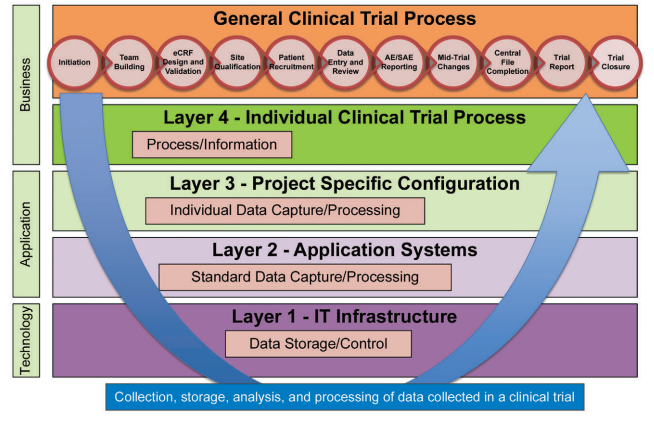
To support the collection, analysis and processing of data collected in a clinical trial, highly specialized tools like an EDC system, CTMS or IVRS/IWRS have been developed and have been in use for years. However, today these tools are no longer stand-alone systems as they have been in the past. These systems have become the building blocks ofan integrated eClinical Platform that supports the efficient conduct of clinical trials (Figure 2.1).
Not all clinical trials will need all systems being part of such an eClinical Platform (e.g., an open-label trial does not require systems that support blinding of trials). For instance, a Phase I trial may require different systems than a Phase IV trial. A Phase I trial may not require an expensive, complex, multi-lingual and web-based EDC system, as Phase I trials are often conducted in only one location with very few users and subjects. Other examples include a subject recruitment database or a barcode reader that may not be necessary in a Phase IV trial.
Additionally, some systems (e.g., a CTMS) will collect and process data from all clinical trials conducted by the organization without further customization while others (e.g., EDC system) may need to be setup and configured for each individual trial based on the protocol requirements.
A further aspect that needs to be considered is potential outsourcing of activities and the usage of SaaS offerings. The resulting eClinical Platform might span across multiple organizations (e.g., the sponsor of the trial), one or more CROs and SaaS vendors (e.g., for an EDC system) and could even include Electronic Health Records (EHR) systems at the investigator sites.
As mentioned in the introduction, a practical and efficient approach for the validation of GCP relevant systems has already been provided in the ISPE Concept Paper 2.
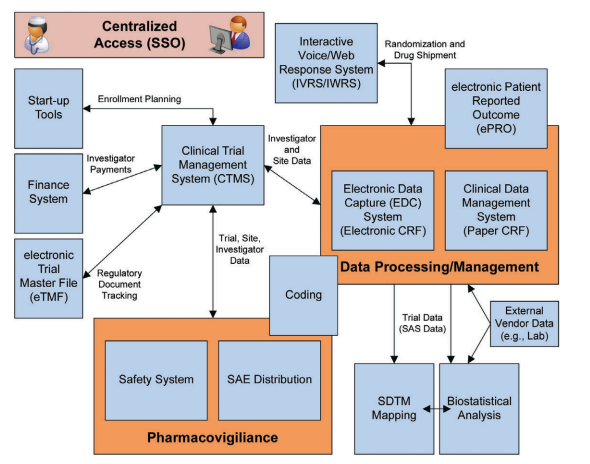
A generic example of an eClinical Platform is provided in Figure 2.2.
Figure 2.2: Example of a Generic eClinical Platform
Definition
Data integrity can be defined as the validity of data and their relationships. For electronic records collected and processed as part of a clinical trial to be trustworthy and reliable, the links between raw data, metadata, and results must not be compromised or broken. Without data integrity, it is not possible to regenerate a previous result of a clinical trial reliably.
Obviously, maintaining data integrity is an important aspect not only for clinical trials and eClinical Platforms. This needs to be addressed throughout the product lifecycle spreading across GMP, GLP, GCP and other GxP areas.
In considering all of these aspects, it becomes obvious that data integrity cannot be ensured by the validation of the individual systems and their point-to-point interfaces alone. A more holistic approach toward validation, including relevant processes, data and quality management is necessary because those systems are acting together across corporate borders and controlled by different quality systems. Similar to validating individual systems following a risk-based approach, the risks of using the eClinical Platform must be identified, assessed and adequately addressed. In addition to the system and study life cycles, this holistic validation approach needs to support the complete data life cycle from the first generation of the data till the end of the retention periods and should include the relevant metadata. To limit the scope of this paper, only the validation aspects of the systems and platforms are investigated. Other techniques and controls need to be in place to assure data integrity along the complete data life cycle.
3 Generic eClinical Platforms vs. Trial-Specific eClinical Platforms
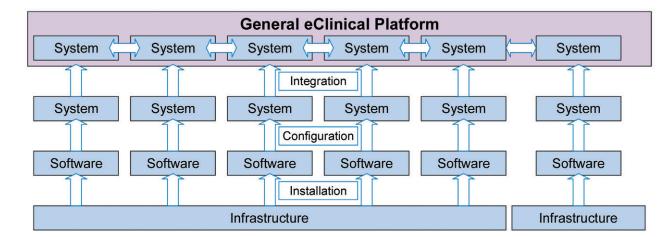
As it would be inefficient to build a full eClinical Platform for every trial, establishing a generic eClinical Platform based on the individual GCP systems is required.
A generic eClinical Platform consists of all potentially required systems for the conduct of a clinical trial (Figure 3.1). These are typically connected via numerous interfaces. Basic functionality and configuration that is required for the majority of clinical trials is included and validated following a risk-based approach.
This generic eClinical Platform also includes all SaaS offerings and systems from strategic partners like CROs that are frequently used for the conduct of clinical trials. The necessary transfer of data between the clinical trial site, CRO and sponsor adds significantly to the complexity of the platform.
Figure 3.1: Building a Generic eClinical Platform
This generic eClinical Platform provides the validated baseline for any trial-specific platform and additional validation activities. This validated baseline enables organizations to establish a “trial-specific” eClinical Platform (Figure 3.2) in a timely manner as only aspects that differ from the baseline need to be validated for the setup and configuration of the trial. Depending on the type and complexity of the clinical trial, this trial-specific eClinical Platform may only contain a subset of the systems offered by the generic eClinical Platform and includes trial-specific setups and configurations of these systems. Furthermore trial-specific requirements may require the development, set-up and/or configuration of trial-specific interfaces within the organization and/or between organizations.
Figure 3.2: Creating a Trial-Specific eClinical Platform
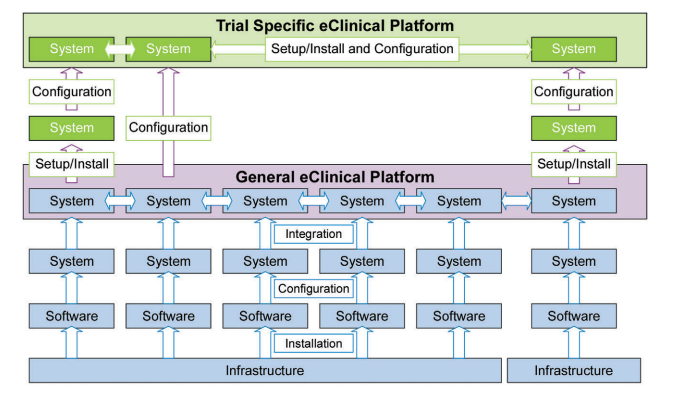
4 The Foundation – Validation of the Individual Systems
The validation of the individual systems forms the foundation of all further quality management activities.
As illustrated in Figure 4.1, the platform life cycle is based on the general requirements that would support a generic clinical trial. Therefore, in order to assess the validation for systems used in the context of a clinical trial an assessment of the general underlying platform life cycle as well as the trial-specific life cycle is necessary.
Figure 4.1: Applicable Life Cycles
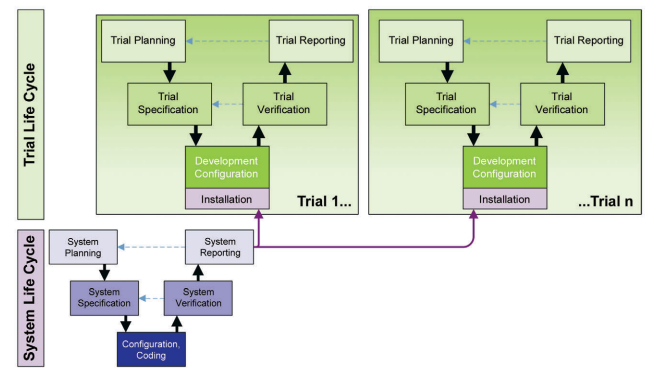
The trial life cycle is based on the trial-specific requirements as determined by the team supporting and executing an individual clinical trial project.
Depending on the type and function of the system, the validation effort may be greater for the platform life cycle than the trial life cycle aspects (e.g., safety systems typically require only very limited setup and configuration to address trial-specific needs as the processing of Serious Adverse Events (SAEs) is quite uniform and regulated in fine detail.). In contrast, the trial-specific setup of an EDC system, including the necessary electronic Case Report Forms (eCRFs), often requires significantly more effort while only basic functionally can be validated as part of the platform life cycle.
However, without the integration with other systems, these individual systems are just isolated building blocks that would not appropriately support the various processes necessary for a clinical trial. Only through integration with other systems can they truly form an eClinical Platform. Obviously any automated transfer of data via interfaces to systems that potentially support a different part of the business process can add risks for the integrity of data as well.
5 Interfaces (Technical Aspects)
Interfaces may be between internal systems or reach across multiple organizations. The setup often includes systems from the sponsor, CRO(s), various suppliers including EDC providers, laboratories, electronic Patient Reported Outcomes (ePRO) providers, logistics and others. Additionally, systems at the investigator site like EHR systems may be included in the eClinical solution.
Interfaces as part of eClinical Platforms can be categorized as:
- Non-Configured Interfaces
- These interfaces do not require any configuration and usually transfer data that are processed as part of every trial. These interfaces would typically be validated as part of the generic eClinical Platform validation.
- Example: CTMS to safety system integrations
- These interfaces do not require any configuration and usually transfer data that are processed as part of every trial. These interfaces would typically be validated as part of the generic eClinical Platform validation.
- Configurable Interfaces
- These interfaces require significant planning, management and control, and usually transfer data that are processed as part of every trial as well as trial-specific data. The validation of these interfaces is often split between the generic eClinical Platform validation (i.e., validation on system level) and the trial-specific eClinical Platform validation (i.e., validation on trial level).
- Example: EDC system to CTMS integrations or EDC system to IVRS/IWRS
- These interfaces require significant planning, management and control, and usually transfer data that are processed as part of every trial as well as trial-specific data. The validation of these interfaces is often split between the generic eClinical Platform validation (i.e., validation on system level) and the trial-specific eClinical Platform validation (i.e., validation on trial level).
- Trial-Specific Custom-Built Interfaces
- These interfaces are custom-built to address a very specific need of the trial, or group of trials, and are unlikely to be used again for future trials. The validation of these interfaces is typically conducted during the trial-specific eClinical Platform validation.
- Example: EDC system to ePRO integrations or partly manual interfaces (e.g., upload of laboratory results)
- These interfaces are custom-built to address a very specific need of the trial, or group of trials, and are unlikely to be used again for future trials. The validation of these interfaces is typically conducted during the trial-specific eClinical Platform validation.
Consideration of who operates and maintains a system is also helpful:
- Interfaces between systems of the same organization
- Interfaces between trusted partners
- Interfaces between one-time/first-time partners
While integrated systems of the same organization are usually very well controlled, the setup, operation and maintenance of interfaces between different organizations are more challenging. This requires well-established communications and sound contractual agreements to address potential differences in the validation approach and agreement on standards to be used. These arrangements are typically more mature between trusted partners that are considered to be reliable, trustworthy and have been utilized and audited several times. Typically, these partners have the status of a preferred supplier/vendor and there is a well-established Service Level Agreement (SLA) in place.
6 Interfaces (Organizational Aspects)
Ensuring the integrity of data collected and managed by a computerized system is essential to support the evaluation of the investigational product and ultimately protect patient safety. As stated in ICH E6 Guideline for Good Clinical Practice 3, “the ultimate responsibility for the quality and integrity of the trial data always resides with the sponsor;” therefore, the sponsor has to establish adequate quality oversight.
The current trend of pharmaceutical companies to outsource significant parts of their clinical trial activities and IT systems often leads to a complex eClinical Platform involving several partners, one or more CROs and several IT providers.
An EMA Reflection Paper4 discusses outsourcing of the eClinical Platform to a third party with strict controls over sponsor access. This could help the sponsor demonstrate that they do not have exclusive control over source documents and/or data.
To ensure the essential quality aspects of a clinical trial, the foundation of the cooperation between these parties must be laid out in contracts and Master Service Agreements (MSA), Statements of Work (SOW) and similar legally binding documents. Including basic quality, organization and communication requirements in these documents is considered a good practice.
These expectations can include:
- Audit frequency or schedules
- Establishing Steering Committees/Governance Boards, etc.
- Expectations for metrics/Key Performance Indicators (KPIs)
- Advance notifications for system changes and/or downtimes
- Direct access to partner systems and/or data
- Details of data transfer (i.e., security, frequency, technology, etc.)
- Usage of electronic or digital signatures
- Details for protection of personal data (e.g., Safe Harbor)
- Change control aspects (e.g., review and approval considerations) for processes, systems, data and trials
- Upgrade/maintenance schedules
- Strict controls over sponsor access to independently hosted data
- Training requirements, especially regulatory
- Non-disclosure agreement(s)
- Escrow agreement
- Notification for subcontracting, including third-party data hosting or international cloud provisioning of data
- Long-term data archival and retention
- Data/system access in the event of partner (or third-party hosting) bankruptcy
- Notifications necessary for privacy and confidentiality breaches
It is recommended that change control aspects are already considered in the contract/MSA, as changes made to systems or processes are likely to break interfaces or to put data integrity at risk.
Change control should include mid-trial changes that mandate technical modifications, as an evaluation of the Tufts Center for the study of drug development clearly demonstrated that protocol amendments and modification are normal and not exceptional 5.
Agreements should outline the types of system changes that must be communicated to the sponsor and which changes require an approval prior to implementation.
Also the usage of electronic signatures and the validity of those, when the data are transferred from one partner to another, need to be carefully considered. It might be necessary to use digital signatures for some data elements to allow verification of the signature at any time, and this may prove problematical after the trial has completed.
Generally, open and honest communication between all involved partners are essential for the success of an eClinical Platform and subsequently the success of the trials conducted. Therefore, not only are contracted items crucial for data integrity throughout the entire trials life cycle, but also the contact between the responsible project’s personnel involved in the trial. Regular meetings (e.g., weekly, biweekly, etc.) during the trial life-time are essential. Furthermore, the participation of all relevant parties, including business, IT and quality is crucial as systems and trial setup are likely to change over time. A good team, managed by project managers from the sponsor and the supplier(s), is a key factor for the success of the trial and maintaining data integrity. Another important aspect is that the supplier(s) fully understand the regulations that the sponsor must comply with, and that they have a vital part in ensuring that compliance is maintained. This includes supporting audits and inspections by the supplier and/or regulators, but also the timely reporting of incidents to the sponsor. The criteria for reporting of incidents may be documented within a contract.
Note: the protection of personal data and patient confidentially as well as the blinding/un-blinding of data pose additional challenges.
However, it also must be understood that the intellectual property of the involved partners must be protected, as some partners may be competitors in the same market. For example, a sponsor may work with several CROs or EDC providers. This should be considered throughout all communication channels when multiple parties are involved.
7 Validation in Different Organizations
Every interface can potentially endanger the data integrity of a trial. Consequently, all interfaces and systems should be assessed and validated following mutually acceptable standards. This is often very difficult to verify, achieve, or establish if the eClinical Platform involves systems validated by different organizations following their own internal standards and procedures.
Terminology is often used inconsistently across various parties and thereby produces additional challenges in communication. A mapping of the individual validation deliverables and terminology to the GAMP® 5 Guide can establish transparency and identify gaps. This can be done as part of regular audits or as part of on-going communications between the involved organizations.
Sponsors may consider establishing a “Quality Committee” with representatives from all strategic/preferred partners to define requirements and develop a common approach. This committee may also ensure that all partners are aware of the most recent changes in relevant regulations. It is noteworthy that recent guidance released by the U.S. Food and Drug Administration (FDA) explicitly states that FDA does not intend to assess the compliance of EHRs with 21 CFR Part 11 6.
8 Dataflow and End-to-End Validation
Aristotle is attributed as saying:
The whole is more than the sum of its parts.
In this context, even if every involved system for every involved partner has been validated to mutually agreed standards and processes, including clearly defined risk-based approaches, this may still not be enough to ensure the integrity of data.
The remaining risk results from the fact that the data are not just being exchanged between two systems or two parties. Some of the data flows through multiple systems, at multiple partners, as illustrated in Figure 8.1.
Figure 8.1: Data Flows in eClinical Platforms
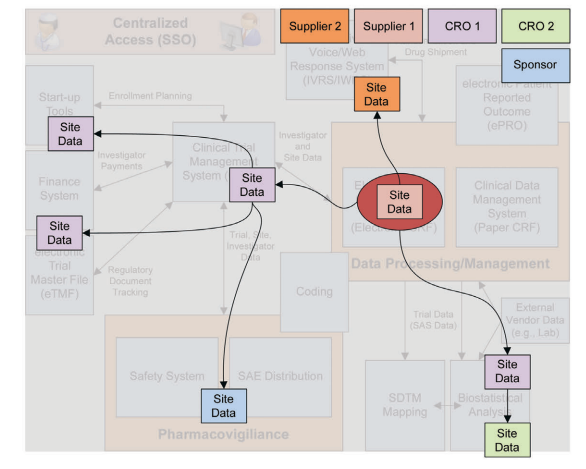
The individual teams responsible for a system are often aware of the immediate partners or systems with which they exchange data. However, most of the time, they are unaware of any further hand-overs or data transfers. Therefore a change in System A, or the processes applicable to System A, may not have an effect on the directly interfaced System B. But it may very well be having an effect on System C that is interfaced with System B.
Obviously this could be avoided if every system would “read” the required data directly from the source. However, this might not be possible for either procedural and/or legal reasons.
An Example:
A trial start-up tool provides site data (i.e., name, contact details, etc.) to the CTMS. At this point in time, these sites have not been “initiated,” as they are imported into the CTMS as “ready for initiation.” Once the initiation is completed, the data are transferred to the EDC system and the sending of login details is triggered.
A change in the way the data are collected in the trial start-up tool (e.g., fax numbers are no longer captured) may not have direct consequences in the CTMS (as the users use other means of communication such as e-mail, etc.); however, it could trigger problems in the EDC system (as it sends the user name via e-mail but the initial password via fax).
Data that are often transferred in such a way includes:
- Investigator data
- Often processed in CTMS, EDC, IVRS/IWRS and financial systems
- Site data
- Often processed in CTMS, EDC, IVRS/IWRS and trial start-up tools
- Safety data
- Often processed in CTMS, EDC, and safety systems
As a consequence, data integrity is at risk.
However, the integrity of the data could be established by an end-to-end verification of the eClinical Platform.
This could be achieved by setting-up a trial in the test environment of the full eClinical Platform. In this setup, all processes changes for the systems, or for the trial, could be tested prior to “release to production” in order to mitigate risk to data integrity. However, this is a rather costly approach.
Alternatively, if data flow could be analyzed along the clinical processes, the risks associated to these data flows could be determined. This would require a much more detailed analysis of the processes, data and records than it is typically done today. Furthermore it would require a Review Board that would be able to carry out extensive impact and risk assessments of each change request.
It must be understood that these activities are not limited to automated data transfers. Quite often data are transferred at specific intervals or at specific milestones of the project by “manual” handovers. An example would be the handover of the locked database content from data management, at a CRO, to the sponsor or other party for further analysis. This is often done via a manual upload to a secure File Transfer Protocol (FTP) site.
9 Summary
With the on-going trend for outsourcing clinical trial activities, the risks to data integrity become higher and more visible.
Tightly integrated eClinical Platforms spanning across multiple organizations have been established over time and should be considered a standard practice. The tightly integrated flow of data between multiple systems requires additional controls to ensure data integrity during the data creation and collection stage of the overall clinical data lifecycle.
Foremost the importance of cross-organizational communication cannot be underestimated. It has become apparent that significantly more effort is required by the sponsor than to just conduct regular audits. Due diligence and regular audits of the supplier(s) are no substitute to active partnership and cooperation.
Contractual agreements are critical to ensure a common understanding of the expectation for each party involved. They need to go significantly beyond the mere financial details and a high level scope of the work to be done. Quality standards and communication aspects should be included at this stage as well.
In addition, a risk-based approach to validation must not only be applied to individual systems, but also must be taken upward to the next level. A holistic risk assessment of the eClinical Platform, including data flows across integrated systems, must be performed with additional controls added as necessary. This can include processing of a trial in a test setup of the platform or end-to-end tests across the eClinical Platform for critical data flows.
The most suitable approach must be determined for each eClinical Platform on an individual basis, as they vary greatly in complexity. As with most systems, the greater the complexity the greater the risks, and the more necessary additional controls are to implement.
While this paper specifically addresses some of the technical and organizational aspects of data integrity, the impact of human error or fraud to the integrity of data is not covered. These aspects require additional thought and should be discussed separately.
11 Abbreviations
| CFR | Code of Federal Regulations |
| CRF | Case Report Form |
| CRO | Contract Research Organization |
| CTMS | Clinical Trial Management System |
| eCRF | electronic Case Report Form |
| EDC | Electronic Data Capture |
| EHR | Electronic Health Record |
| EMA | European Medicines Agency |
| ePRO | electronic Patient Reported Outcome |
| eTMF | electronic Trial Master File FDA Food and Drug Administration (US) |
| FTP | File Transfer Protocol GAMP Good Automated Manufacturing Practice |
| GCP | Good Clinical Practice GLP Good Laboratory Practice |
| GMP | Good Manufacturing Practice GxP Good X Practice (X can mean: Clinical, Laboratory, Manufacturing, Pharmaceutical, etc.) |
| ICH | International Conference on Harmonization |
| IMP | Investigational Medical Product |
| IT | Information Technologies |
| IVRS/IWRS | Interactive Voice Response System/Interactive Web Response System |
| SaaS | Software as a Service |
| SAE | Serious Adverse Event |
| SDTM | Study Data Tabulation Model |
| SLA | Service Level Agreement |
Limitation of Liability
In no event shall ISPE or any of its affiliates, or the officers, directors, employees, members, or agents of each of them, or the authors, be liable for any damages of any kind, including without limitation any special, incidental, indirect, or consequential damages, whether or not advised of the possibility of such damages, and on any theory of liability whatsoever, arising out of or in connection with the use of this information.
© Copyright ISPE 2014. All rights reserved.
No part of this document may be reproduced or copied in any form or by any means – graphic, electronic, or mechanical, including photocopying, taping, or information storage and retrieval systems – without written permission of ISPE.
All trademarks used are acknowledged.
Acknowledgements
By the ISPE GAMP Community of Practice
This concept paper was written and reviewed by members of the Global ISPE R&D and Clinical Systems Special Interest Group (SIG) the ISPE GAMP Community of Practice (COP).
SIG Chairs
Oliver Herrmann, Q-FINITY Quality Management, Germany
Eric Staib, ReSearch Pharmaceutical Services, Inc., USA
Document Authors
Frank Henrichmann, PAREXEL International GmbH, Germany
Marina Mangold, Alcedis GmbHm, Germany
Oliver Herrmann, Q-FINITY Quality Management, Germany
Dieter Wachtmann, PAREXEL International GmbH, Germany
Reviewers
Rachel Adler, Johnson & Johnson, USA
Aneta Blazevska, Abbott GmbH & Co. KG, Germany
Tamara Dehnhardt, B.Braun Melsungen AG, Germany
Jenny Gebhardt, Q-FINITY Quality Management, Germany
Ina Helm, Alcedis GmbH, Germany
Corinne Liabeuf, Novartis Pharma AG, Switzerland
Eric Staib, ReSearch Pharmaceutical Services, Inc., USA
Yvonne Rollinger, OmniComm Europe GmbH, Germany
Maximilian Stroebe, Novartis Vaccines and Diagnostics, Netherlands
Thanabalan Subramanian, Roche Products Ltd., United Kingdom
Frank Woods, GlaxoSmithKline, United Kingdom
Craig Williams, Integrity Solutions Limited, United Kingdom
Regulatory Input and Review
Christa Färber, Trade and Industrial Agency of State of Lower Saxony – Agency Hannover, Germany
Special thanks to reviewers from the Data Integrity SIG:
John Avellanet, Cerulean Associates LLC, USA
Lorrie Schuessler, GlaxoSmithKline, USA
Particular thanks go to the following for their support of this Concept Paper:
Chris Clark, Napp Pharmaceuticals Ltd., United Kingdom
Jonathan Helfgott, CDRH FDA, USA
Arhur (Randy) Perez, Novartis Pharmaceuticals, USA
Michael Rutherford, Eli Lilly and Company, USA
Siôn Wyn, Conformity Ltd., United Kingdom


