Process Validation Lifecycle Implementation for Existing (“Legacy”) Products

This Discussion Paper examines some of the topics and challenges related to the implementation of current Good Manufacturing Practice (“cGMP”) Process Validation (PV) lifecycle concepts in the management of Quality Systems for existing (“legacy”) commercial products. The paper aims to identify common issues related to the application of lifecycle principles in a legacy product context, and to discuss potential responses to various scenarios that may be encountered.
1 Introduction
This Discussion Paper examines some of the topics and challenges related to the implementation of current Good Manufacturing Practice (“cGMP”) Process Validation (PV) lifecycle concepts in the management of Quality Systems for existing (“legacy”) commercial products. The paper aims to identify common issues related to the application of lifecycle principles in a legacy product context, and to discuss potential responses to various scenarios that may be encountered.
Among the topics to be discussed:
- Current Validation Lifecycle expectations for legacy products
- Strategies for assessing and prioritizing requirements for legacy process/process validation
- remediation
- Expectations for revalidating a modified legacy process
- Expectations for revalidating an unmodified legacy process based on existing PV “gaps”
This Discussion Paper is focused on the challenges of implementing a current process validation lifecycle approach in the quality management of a set of products initially validated and commercialized prior to the formal introduction of the current lifecycle architecture. This includes cases where the process was not necessarily developed, characterized, and/or documented in accordance with current concepts (e.g., as described by the current version of ICH Q8(R2) 1 such as “Quality by Design” (QbD)).
Regarding the legacy product processes that it addresses, the assumptions of this paper are as follows:
- Products were previously validated, are currently being marketed commercially, and are typically in the lifecycle stage referred to in this paper as Ongoing Process Verification (OPV), (also known as Continued Process Verification (CPV)), or ongoing monitoring phase
- Original or previous process validation(s) were conducted in compliance with predicate GMP rules current at the time of validation
- A Pharmaceutical Quality System (PQS) consistent with ICH Q102is in place at the manufacturer
The intended scope of this paper includes both Active Pharmaceutical Ingredients (APIs) (Drug Substances) and Drug Products. It also includes both large and small molecule products, and all routes of synthesis and dosage forms. The principles and practices described and discussed are generally applicable, and are intended to comply with understood and accepted international Good Manufacturing Practice (GMP) expectations for pharmaceutical process validations. Products made by, or for, Third Party Manufacturers (TPMs) and/or Contract Manufacturing Organizations (CMOs) are also included.
This paper focuses on issues related to establishing and/or maintaining the “validated state” for legacy products for which some current lifecycle concepts may not have been formally established and implemented.
Organizational strategies will be presented for the prioritization of remediation of legacy product validation lifecycles based on:
- Product risk (dosage form, criticality of clinical effect, etc.)
- Product history (available data, quality history, manufacturing experience)
- Prioritization of products, for which gap closures may be needed
This paper is not intended to be a discussion of the lifecycle validation approach per se, as numerous other articles and information sources exist on that topic. It is not intended to examine issues or practices around process development, technology transfer, qualification of facilities and systems, planned changes to existing products or supporting process validations (such as validation of cleaning or analytical methods). It does, however, consider such topics where they intersect with the general intent of monitoring and maintenance of the validated state, and the actions taken in response to quality data signals to maintain that state.
In addition, this paper does not discuss the mechanics of OPV (such as determining which parameters and attributes to track, setting control/action limits, frequency of process analysis, etc.), as these topics are covered in detail in another ISPE PQLI PV Group Discussion Paper: “Stage 3 – Process Validation: Applying Continued Process Verification Expectations to New and Existing Products” 3.
2 Background
Updated process validation guidance documents from the US FDA, EU, ASEAN, and other regional regulatory authorities have described similar 3-phase validation lifecycles, based on process knowledge and quality risk management in alignment with ICH Q8(R2)1/ICH Q9 4/ICH Q10 2. The Validation Lifecycle discussed in this paper consists of three “stages”, with the starting point of this paper being products that are currently validated and being marketed commercially (in the OPV stage below). In the terminology of the Process Validation Guidelines 5, 6, such products are referred to as “legacy products”. The lifecycle stages referred to in this paper are:
- Process Design: The commercial manufacturing process is defined during this stage based on knowledge gained through development and scale-up activities.
- Process Qualification: During this stage, the process design is evaluated to determine if the process, along with supporting equipment and facilities, is capable of reproducible commercial manufacturing. This typically includes manufacture of the initial validation lots (or Process Performance Qualification (PPQ) lots).
- Ongoing Process Verification: This is post-commercialization and ongoing assurance during routine production that the process remains in a state of control. OPV is actually a two-phase stage, with initial monitoring of new or redesigned products being monitored more intensively based on less historical data and process experience, with more reduced levels of monitoring as statistical confidence builds. The initial phase of OPV is often referred to as “Enhanced” OPV, while the normalized sampling, periodically adjusted in response to the voice of the process, is referred to as “routine”.
Prioritization of perceived gaps, generally, should precede programmatic efforts at remediation. In some instances, a comprehensive assessment of the product may already be available as an output from the Annual Product Review, OPV (if implemented), past site inspection readiness efforts, or recent audits. For such instances, these assessments may be used to support the gap/risk assessment process accordingly. The associated output from the previous assessment should be formally documented and attached as supporting documentation for this protocol.
In all cases for the recommendations and contents of this paper, there will be the intent to conform to the two primary principles of ICH Q9 “Quality Risk Management” 4:
- The evaluation of the risk to quality should be based on scientific knowledge and ultimately link to the protection of the patient.
- The level of effort, formality, and documentation of the quality risk management process should be commensurate with the level of risk.
The flowchart in Figure 1 depicts a two-part “decision-tree”; the top of the chart illustrating the organizational evaluation, followed by an example of a process-specific evaluation with potential options for responses to various scenarios.
Figure 1: Lifecycle Implementation Decision Path – Where to Start?
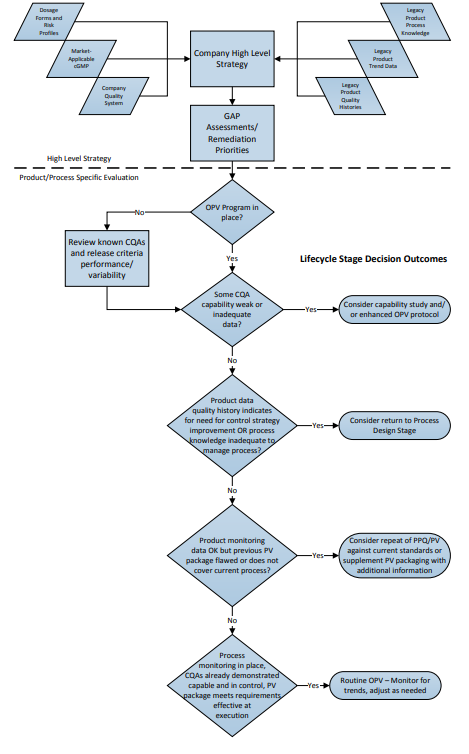
3 Organizational Evaluation of Legacy Validation Programs
Firms with significant numbers of legacy products must make programmatic and strategic decisions regarding the current validated status of each of those products. Evaluations should be based on the ability to demonstrate a state of process control at any time. The validated state of each commercial process depends primarily on these factors:
- Is there an ongoing monitoring (e.g., OPV) program in place for each product that monitors the appropriate attributes and variables, and provides sufficient data to demonstrate an ongoing state of control (i.e., can the effectiveness of risk control strategies be demonstrated through the quality data)?
- Are the outcomes of the ongoing monitoring program reviewed on a timely basis to assure control with appropriate actions and adjustments as indicated by quality data signals?
- Is there sufficient available documented process knowledge (rationalized Critical Quality Attributes (CQAs) and Critical Process Parameters (CPPs), development reports, process justification studies, technical analysis, process experience, etc.) to provide adequate rationale for decisions made regarding changes to the process, investigate nonconforming lots, and otherwise to maintain the state of control of the process?
- Has effective change management been in place since the original or most recent full revalidation, and does the existing PV documentation (protocols, reports, etc.) accurately represent the current process?
- Does the available PV documentation meet GMP regulatory and company policy requirements effective at time of PV?
If a firm’s quality system assures that the above questions can be answered positively for its legacy products, then the product validation lifecycle is effectively already in place, and routine monitoring may continue, adjusting levels of sampling and testing as indicated by events and trends over time.
For some firms, one or more of these questions may represent a gap in quality system procedures or requirements, or there may be “gaps” in documentation for one or more products, which indicate a need for additional remediation. Such actions may be required before, or in addition to, simply implementing or maintaining an ongoing monitoring program.
3.1 Lifecycle Prerequisites – Quality System Readiness
Prior to the programmatic implementation of the process validation lifecycle for a group or set of legacy products, the company and/or manufacturing site need to ensure that they are prepared for lifecycle implementation with the necessary quality system infrastructure and work culture. A support foundation should be embedded within documentation, training, and other quality systems. A summary in Table 1 of this Discussion Paper provides some of the items for consideration when evaluating legacy PV documentation for compliance adequacy, with some specific requirements for biological and aseptic processes. Some current expectations for quality system management of both new and legacy products include:
- Corporate policies and procedures (Quality Management Systems) need to set requirements for the lifecycle and also allow for the flexibility to serve and compete in local markets for companies that operate in multiple (global) regulatory environments.
- Standard Operating Procedures (SOPs) defining site, departmental, and individual roles and responsibilities should be in place or created to assure a routine focus on product/process quality management. Considerations include:
- technical product ownership and process expertise
- statistical requirements, where appropriate, for assurance of control
- data handling and review
- quality oversight and management review
- Company/Site SOPs, validation plans, and quality systems (such as deviation management, change control and periodic product review) may need to be updated to incorporate a lifecycle
- approach.
- While manual compilation and analysis of process data is possible, automated systems to handle the data trending requirements of OPV greatly facilitate tracking and trending, and in cases of high product volumes, may be a practical necessity.
4 Organizational or Site Assessment
Following necessary updates to corporate policies and procedures, individual manufacturing sites can start to develop detailed site-specific plans. Since the process of lifecycle implementation is not immediate (especially for multi-product facilities), a multi-phase, multi-year approach may be required. The plan should be developed to help bridge the gap between the current state and the desired future state. The plan should include:
- a schedule for implementation for any identified remediation activities
- A prioritization of products with consideration focused on patient impact. Factors may include:
- volume of product in the market
- number of patients served
- product Criticality (i.e., is the product a lifesaving therapy)
- products for which there may be shortages, or which are unique in meeting specific medical needs
- regulatory Authority
- process quality history (% batches rejected, total complaints received separated by critical, major and minor, etc.)
- total GMP deviations (separated by critical, major and minor)
- planned process changes
- total change controls opened since last PV
- manual / high risk control strategies
- status of the existing PV documentation package for a given product
- the amount of ongoing monitoring data (if any) currently collected for given product
- a procedure on updating the plan based on new knowledge or events
- Handling of products developed to different standards (i.e., some will have formal CQAs and CPPs documented while others may not)
4.1 Product/Process Specific Evaluation and Remediation Options
Once site-specific or organizational priorities are established as above, individual process validation document packages can be gap and risk assessed for alignment with the revised quality system expectations and appropriate remediation identified.
Figure 2
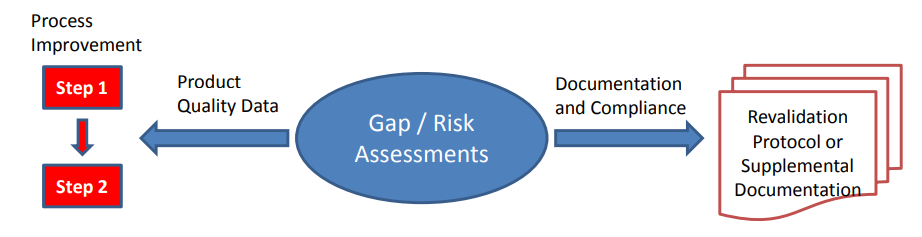
Depending on the nature of any gaps or risks identified for individual products or processes, remediation plans can be broadly divided into two strategies; remediation plans that may require actual process changes to effect improvements (changes including risk control strategy improvements) and those that do not, being documentation or compliance in nature, and often remediable without actual process change. Process changes should be implemented through change management systems with associated risk management activity. They usually require revalidation in order to assure that critical impact of changes have been identified and controlled.
5 Current PV Program Requirements
| Item | Considerations/Expectations |
|---|---|
| General Processes | |
| Process Design |
|
Hold Times for Process Intermediates |
|
| Starting Materials |
|
Materials of Product Contact |
|
| Filter Validation |
|
Shipping/ Transportation Qualification |
|
Processing Solutions (e.g., Buffers) Preparation and Shelf Life Qualified |
|
Impurities Clearance (Impurities removal/clearance) |
|
| Chromatography Resin and UF Membrane re-use |
|
| Biological Processes | |
| Cell and Virus Banks |
|
Viral clearance/ inactivation (from the process stream) |
|
| Aseptic Processes | |
| Filter Validation |
|
| Media Simulation |
|
| Container Closure System Qualification |
|
| Bulk Container Integrity |
|
5.1 Potential PV Documentation Gaps and Recommendations
Table 2 outlines some potential PV package gaps for individual processes.
| If... | Then... | And... |
|---|---|---|
| There is not an OPV program in place with monitoring and trending (where appropriate) of known CQAs and CPPs... | ...the process may or may not be operating in a state of control and the adequacy of the process control strategy cannot be assured... | ... attributes and parameters to be monitored should be established and the historical performance of the process for the chosen attributes and parameters analyzed using release and in-process data. |
| The available quality history for a product manufacturing process shows repeated events or poor capability – or process knowledge is insufficient to effectively manage... | ...the process may be poorly controlled (control strategy needs improvement) or may need some level of actual process redesign... | ...the applicable lifecycle stage could be design stage, followed by a review of equipment/system qualifications (e.g., Stage 2a) to assure coverage of any new or modified parameters, and then “revalidation” under a rationalized PPQ/PV protocol. |
| Quality history is generally good but some quality attributes are weak (e.g., CpK/PpK) or are not currently monitored/ trended... | ...consider whether a weak process capability is likely to be improved with more data (such as in cases of limited sample sizes or data) or whether the process needs a specific improvement (as above)... | ...an OPV enhanced monitoring protocol or study may clarify whether or not controls are adequate. Enhanced sampling and testing would be focused on the weak or missing attributes or parameters. |
| Quality history is good and critical attributes and parameters have been shown to be well controlled, but available PV documentation is noncompliant with GMP predicate rule and/or no longer represents current process, or may simply be too old and replacement is desired... | ...no process changes are indicated but a repeat or supplementation of PPQ, “modernized” to include rationales for number of lots, sampling plans, and inter and intra batch variation criteria may be appropriate... | ...the process qualification PPQ need not require subsequent enhanced sampling since process was capable and not changed, and the lifecycle may resume post-PPQ in routine OPV. |
| Change control has been in place, process quality history is good, critical attributes and parameters are well controlled, and an adequate ongoing monitoring program is in place. Available PV is compliant and represents the current manufacturing process... | ...the process is in a state of current validation... | ...routine ongoing monitoring can be continued, monitoring for trends and adjusting sampling plans if and as indicated by ongoing data. |
5.2 Remediation of Gaps – What to Fix?
A frequent “gap” found in product lifecycle documentation for legacy products is poor or missing development documentation. Many legacy products predate current global regulatory expectations established by ICH Q8(R2) 1 and the harmonized definitions of CQAs and CPPs established in that document. A common question/discussion point, that this gap may raise, is around the need to “reverse-engineer” an existing process that lacks one or more of the elements of current pharmaceutical development documentation. It is recommended by this paper that a firm is not expected to perform missed or missing aspects of “development” work, but should instead supplement related documentation so as to “close” these “gaps”, for established and well-controlled legacy processes, with a few understood caveats.
Regardless of whether or not formal CQAs and CPPs for a given process have been so designated, in order to successfully manage the product lifecycle in the long term, it is necessary to understand and document understanding of likely interactions and links between parameters and product attributes. For most legacy products and product types, CQAs and associated CPPs can be established based on current and historical process and product data and information, as well as the characteristic attributes of the dosage form or product class. Discovery and development through actual laboratory scale experimentation based on (for example) Design of Experiments (DoE) is typically not necessary for legacy products. That said, a documented understanding and identification of product quality attributes directly related to drug safety and efficacy, as well as the process parameters that need to be controlled to assure those attributes meet specifications, is necessary in order to demonstrate fully the adequacy of the process control strategy and to validate the process.
Tracking and trending of release and critical in-process testing results may be sufficient to demonstrate process control. Well-controlled legacy processes, generally, do not require additional characterization unless changes are required or until OPV reveals a need for improved controls. In such instances, risk assessments can be useful in determining the relative criticality of parameters, materials, or other items being modified.
Actual deficiencies in a control strategy as reflected in quality-stream data (e.g., test data, complaints, and process deviations) may indicate that additional work is necessary in order to understand and improve the control strategy. It is important to remember that such process improvement scenarios do not drive revalidation per se, but rather are the triggers for process control improvements, only then followed by revalidation (PPQ and perhaps enhanced ongoing monitoring) in order to establish effectiveness of the actions taken.
A final category of “gaps” to be considered may be legacy PV packages that do not conform to predicate GMP regulations or company quality policies effective at time of execution, or have other significant compliance issues. Such documentation may need to be remediated through repeating or supplementing the initial validation (e.g., PPQ), simply to improve the available GMP evidence of validation, as opposed to actions taken to modify or improve the process or control strategy. While a revalidation utilizing PPQ plans and protocols meeting current expectations for the market(s) in question may be the recommendation in such cases, a subsequent period of enhanced monitoring may not be required since the process itself likely has not been changed.
These attributes may or may not have been formally identified as “CQAs”, but such labels are unimportant. The question is; how critical is a given parameter or attribute to patient safety and product efficacy and is that rationale documented? In the absence of other process knowledge, release specifications can be assumed to be “critical” as a conservative method of attribute selection. Using that paradigm, process parameters that must be controlled to assure that these release specifications are met can be assumed to be CPPs.
5.3 Ongoing Process Verification and Annual Reviews
With capable process performance and in the absence of compliance gaps, it may be feasible to initiate product lifecycle management simply by trending critical manufacturing batch data on an ongoing basis as part of a routine OPV program. Ongoing monitoring continues until the product in question is modified, improved, or is discontinued. The level of sampling, testing, and other monitoring activities should be periodically reviewed and adjusted, as indicated by the quality data.
A common question for those establishing OPV for the first time is in regard to the relationship between OPV and Annual Product Review (APR) – referred as Product Quality Review (PQR) in the EU. There can be confusion over whether there is redundancy between these two related but separate activities. In the US, 21 CFR Part 211.180(e) 7requires that firms maintain written records of data for evaluating at least annually, the need for changes or improvements to specifications, manufacturing or control procedures. Included in “...the quality standards for each drug product...” and “...drug product specifications or manufacturing or control procedures” is the results of statistical analysis (as appropriate) of process capability supporting a statistically significant confidence statement.
FDA has stated in numerous forums that complying with 21 CFR 211.180(e) 7 does not necessarily satisfy expectations for ongoing monitoring. That is based on the annual timing of the required report. Certainly, modern quality science does not advocate simply a once-a-year review of how a process is performing. The regulatory intent has always been that a much more timely and effective review and reaction to process data is taking place. That review is then documented on at least an annual basis. The emergence of the lifecycle concept simply provides detail around what the regulators intended all along – that pharma and biopharma companies (like almost all modern industries) use the well-established tools of Statistical Process Control (SPC), as appropriate, to evaluate their level of process control and react and correct as needed to assure a constant supply of high quality medicines.
Legacy product firms, with well-established processes, operating under control (including change control) and that have ongoing monitoring programs that are scientifically and statistically (as appropriate) valid, are unlikely to have to change their practices significantly to comply with the Validation Lifecycle concept.
5.4 Revalidation of Legacy Products
When revalidation of a legacy product is required, either due to a process change or as a compliance/documentation improvement, there are a number of issues beyond those raised by a process change. Table 3 proposes some responses based on the principles of the lifecycle approach to some common questions.
| FAQ or Technical Issue | This Paper’s Response |
|---|---|
| Is revalidation required for a legacy product without substantial development documentation? What are the expectations for the Process Design Stage? | Need to have sufficient understanding of interrelationships between materials and parameters and their impact on CQAs to develop/maintain an effective risk control strategy. If operational experience and quality history are adequate, that information can serve as process knowledge basis; leverage existing documentation, data, and understanding. Significant changes to process design should be supported on their own merits by technical evidence meeting current standards, regardless of lack of earlier development. No expectation for firms to “reverse engineer” development documents for established processes, currently operating in a state of process control, simply to represent “Process Design” if CQAs and CPPs can be justified using existing data and information. |
| Is revalidation expected for legacy products based on existing 3-lotlegacy process validations, without justification for number of lots and sampling plans? | The expectation is to demonstrate process control. If a robust OPV sampling and monitoring program is in place, the sampling plan of a PV protocol from many batches ago becomes irrelevant. The “voice of the process” will confirm the control strategy or indicate the need for improvement. If ongoing monitoring data is weak or missing, the existing PV data may be the only direct evidence of process uniformity and control. However, simply revalidating with the intent to test more lots will not ensure that significant sources of process variation are captured. Implementation of an enhanced ongoing monitoring program will either confirm, complement, or contradict the conclusions derived from existing PV protocol data in respect to process stability, process capability, and product acceptability. |
| How do you justify the number of lots and sampling plans for a legacy product PPQ? | The same way that it is done for a new process: based on process knowledge regarding the inherent variability of a given process, and the assessed risks and strategies to maintain control of that variability. A key difference, however, for legacy product is the significant advantage of having empirical experience and lot data. Assuming even reasonable post-marketing quality data monitoring, a significant amount of information may be available to support decisions regarding the most variable quality attributes and/or any areas of control strategy weakness. Focus should be on sampling and testing on these most variable attributes and 3 lots should be considered a general minimum (in order to demonstrate reproducibility and consistency) and the justification aimed at explaining why not more (lack of extraordinary variability, etc.). NOTE: For changes to an existing process less than 3 lots may be justified if risk is determined to be low. |
| What if there are no defined CQAs or CPPs? Can you use release specifications as acceptance criteria? | All attributes with release criteria (i.e., registered specifications or in-process limits) are typically included in the enhanced data collection and data evaluation plan during PPQ (PV). Process parameters that need to be controlled to assure that these release specifications are met can be assumed to be CPPs. Formal designation of CQAs or CPPs for legacy products is optional. That said, release specification ranges might not be appropriate for direct use as acceptance criteria. The acceptance criteria and sampling used during PPQ for legacy products should allow for appropriate demonstration of within batch control (i.e., intra-batch control),as well as batch acceptance criteria that provide confidence that the batch meets the product specification requirements. |
6 Summary and Conclusions
The5.4 Revalidation of Legacy Products lifecycle approach to PV applies to legacy products and the framework can be instrumental in assessment of products and processes and in addressing gaps. The goal of applying the lifecycle approach to legacy products is the same as for new products; to understand the impact that the various sources of process variability have on product quality, to have an effective control strategy to manage that variation, and to generate documented evidence verifying that the applicable control strategy assures robust process control.
Products do not need to be revalidated for the sake of complying with the “lifecycle approach” in the absence of missing data or control issues. Legacy products are likely to enter the lifecycle in the OPV stage, and any additional remediation actions are dictated by evaluation and prioritization of deficiencies related to product and process performance and/or monitoring plan deficiencies. The level of routine sampling and testing for those CQAs and CPPs that demonstrate product uniformity and compliance with label claim should be evaluated to assure that an appropriate statement of quality can be made for each batch.
Identified gaps may lead to additional PV design activity to improve understanding and control followed by subsequent PV activities to begin verifying the acceptability of the control strategy changes and associated monitoring plan changes, but in general there is no expectation that a firm “reverse engineer” well-controlled legacy processes with documented CQAs and CPPs to fill in gaps in developmental data that are not adversely impacting the process control strategy.
Limitation of Liability
From the issuance of ICH Q8, Q9, and Q10 and the FDA Process Validation Guidance as well as subsequent guidance from various regulatory bodies, the ways of working in process validation have shifted dramatically to a lifecycle approach where decisions are based on product and process knowledge. The guidance that has been developed tends to be based upon new product introductions, but there are expectations for products which were validated and commercialized before this new guidance. This discussion paper examines some of the topics and challenges related to the implementation of current GMP process validation lifecycle concepts in the management of Quality Systems for existing products. This paper will discuss topics such as Current Validation Lifecycle expectations for legacy products, strategies for assessing and prioritizing requirements for legacy process/process validation remediation, expectations for revalidating a modified legacy process, and expectations for revalidating an unmodified legacy process based on existing PV “gaps”.
The paper may be modified or expanded sometime in the future to reflect additional input.
Please direct all feedback to pvpapers@ispe.org.
Acknowledgements
By Authors: David Dolgin (PSC Biotech), David Hughes (Sandoz), Matthew McMenamin (GSK), Parab Parkash BMS), Domenico Schiavone (Fresenius-Kabi), Chris Stevenson (Baxter)


