Embracing the Unknown: QRM Strategies for Cell and Gene Therapies Facilities

The pharmaceutical landscape is rapidly evolving, and cell and gene therapies (C>) are at the forefront of this transformation. These therapies are revolutionizing how we approach patient care, particularly in the realm of personalized medicine. However, this innovation has also introduced challenges, especially when establishing new manufacturing facilities.
One such challenge is qualifying equipment and utilities for drug manufacturing when critical quality attributes (CQAs) and the associated critical process parameters (CPPs) are not clearly defined and the relationship between quality attributes and process parameters is not yet fully understood due to an early development stage. This is a common scenario in multipurpose and new modalities facilities where future products are unknown. This raises the question: How can we ensure product quality and patient safety in such a dynamic and uncertain environment?
What Are New Modalities?
Traditional drug platforms such as small-molecule therapies and monoclonal antibodies are now established in the health care industry; new modalities that have emerged in the past 20 years include C>s, RNA drugs, and complex biologics.1 C>s cover a broad spectrum of therapies (see Figure 1).
Cell-Based Therapies
For autologous therapies, cells are taken from the patient and genetically altered outside of the human body (in vitro) and then usually cultivated. Those cells are then reintroduced into the patient’s body. These products are characterized by a one-patient, one-batch relationship (single-patient therapy). The facilities are for small batch sizes and more “lab type” with small size and tabletop equipment. Also, the manufacturing facilities can be distributed (e.g., in hospitals).2
![Figure 1: Defi nition and pipeline snapshot by modality [2].](/sites/default/files/2024-09/0924_PE_SO_Vid_01.jpg)
![Figure 2: Relationship between CQAs, CPPs, CAs, CDEs, and associated testing [6].](/sites/default/files/2024-09/0924_PE_SO_Vid_02.jpg)
For allogeneic therapies, cells are derived from human donors or other cell lines (such as induced pluripotent stem cells), with or without in vitro genetic modification, and the final cell therapy can be used to treat many different patients. Facilities for such therapeutic products consist of several small-scale operations (cell isolation, centrifugation, expansion, etc.). After cell preparation, the production process at scale starts as known from biotechnology product manufacturing.
Gene Therapies
For gene therapies, functioning genetic material is delivered, for example, with a viral vector into the human body to treat or prevent diseases. Facilities for such therapeutic products allow for bigger batch sizes and one batch is used for multiple patients. These kinds of facilities can consist of equipment or systems known from mature modalities for producing drug substances and equipment for the formulation, known as fill and finish.3
Unknown Product and Process Requirements
Pharmaceutical companies need to get ready for their clinical/commercial manufacturing at an early stage of development when it is not clear which product or therapy will make it successfully out of the development pipeline.
A platform approach can help companies target multiple modalities. Platform facilities are adaptable to various processes and products. This is to support the production of new modality products or therapies and offer flexibility and a faster response to pipeline demands and changing market demands.
System, product, or process requirements are often not fully established, particularly in cases like ongoing development, multiproduct/platform approaches, and contract manufacturing facilities, as well as research and clinical manufacturing settings. With new modalities, further challenges arise with new or nonstandard equipment. In these instances, a foundational design is often based on a set of general process requirements or performance criteria, which are shaped by both the user’s needs and regulatory standards, such as ISO 5/Grade A conditions for aseptic filling with unidirectional airflow.
Identifying Critical Parameters
ICH Q8(R1), ASTM E2500-20, and the ISPE Baseline® Guide Volume 5: Commissioning and Qualification (Second Edition) describe how quality aspects from pharmaceutical development, or defined CQAs, are translated into the requirements of a manufacturing process, with the associated CPPs, to consistently deliver quality products.4, 5, 6 Those requirements are an integral part of the quality risk management (QRM) application, which forms the basis for commissioning and qualification (C&Q).
ICH Q8(R1), ASTM E2500-20, and the C&Q Baseline® Guide categorize these aspects into:
- CQA: A physical, chemical, biological, or microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired product quality4
- CPP: A process parameter whose variability has an impact on a critical quality attribute and therefore should be monitored or controlled to ensure the process produces the desired quality4
- Critical aspects (CAs): Functions, features, abilities, and performance characteristics necessary for the manufacturing process and systems to ensure consistent product quality and patient safety5
- Critical design element (CDE): Design functions or features that are necessary to consistently manufacture products with the desired quality attributes6
The C&Q Baseline® Guide illustrates the relationship between these aspects using automated temperature control and monitoring of process steps as an example (see Figure 2).
From Unknown CQAs and CPPs to Generic System Quality Attributes and System Process Parameters
In scenarios where traditional CQAs and CPPs are not defined, it may be beneficial to use a more generic term than CQA or CPP. The concept of system quality attributes (SQAs) and the associated system process parameters (SPPs) offers a strategic alternative. SQAs are the physical, chemical, biological, or microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired system product quality. The system product is the output from that system. SPPs are the parameters whose variability has an impact on an SQA and therefore should be monitored or controlled to ensure the process produces the desired quality.
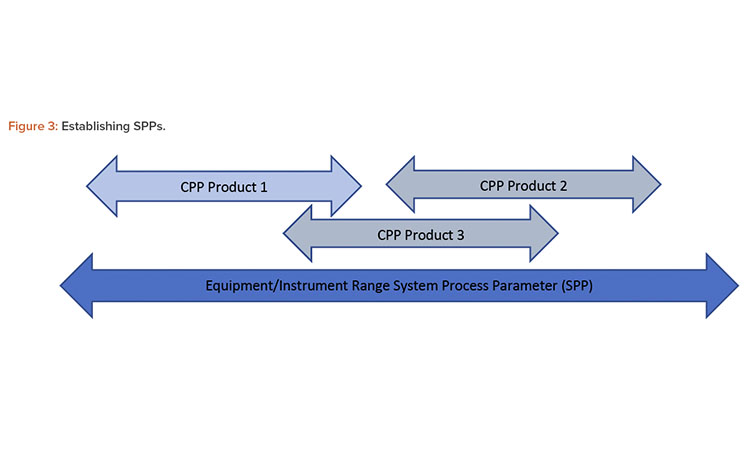
The SPPs represent the full instrument and equipment range and are not specific to one product or manufacturing process. SQAs and SPPs are derived by gathering and analyzing a range of information, including data from existing documentation and the insights of subject matter experts (SMEs). An alternative terminology would be equipment quality attribute and equipment process parameter.
A foundational design is often based on a set of general process requirements or performance criteria, which are shaped by both the user’s needs and regulatory standards, such as ISO 5/Grade A conditions for aseptic filling with unidirectional airflow.
Also, where the equipment maybe be used for multiple products, there may be variation in the specific CQAs and CPPs for the products. The use of the broader terms SQA and SPP, which can encompass all the CQAs for the products that will be processed on the equipment, is then beneficial. This can also help explain why there may be more attributes or parameters used for equipment qualification than there are in a product filing, for example.
Figure 3 illustrates the establishment of SPPs where development and process SMEs provide expected or likely CPPs while going through different steps of a system risk assessment (SRA). In the absence of a CPP system, qualification will cover the full equipment/instrument range.
System Risk Assessment
The process of identifying and validating SQAs and SPPs is greatly enhanced by using risk assessment methodologies, as detailed in the C&Q Baseline® Guide. This framework provides a structured approach to assess and document the SQAs and associated SPPs, assessing the potential impact of each SPP on the overall process and product quality. This approach is especially valuable when we deal with new technology or with complex systems, which is a challenge often associated with C> facilities.
Developing an understanding of the operational sequence of the process helps identify the SQAs and SPPs. It also helps build the knowledge of processes, systems, and equipment. By employing this method, manufacturers can ensure that even in the absence of traditional CPPs, the manufacturing process remains robust, controlled, and capable of producing high-quality products. This adaptation not only aligns with regulatory expectations but also facilitates a more agile and responsive manufacturing environment. The SRA methodology is demonstrated in the following example. We have picked this example because everyone might know washers and therefore can easily follow the concept.
As shown in Table 1, the SQA is general in the example—as the C&Q of the washer confirms that the system delivers the specified requirements. Cycle development and cleaning validation will confirm that the system is fit for its intended use. The same strategy can be used for process equipment with general parameters applied; these may apply to all utilization of the system or be specific to individual products.
The SRA considers the risks to the output of the system based on the proposed design and anticipated controls (design and procedural). The SRA then determines if the controls are adequate to provide an acceptable risk profile for that system.
Efficient Use and Application of the SRA
The process suggested to ensure efficiency is as follows. First, develop the initial draft of the SRA with a small group—system/process SME, facilitator, and quality; this makes the process quick. Then review the draft with a larger team, including operations and maintenance. This provides the benefits of letting the site staff get an understanding of the equipment and associated process early and ensuring that their localized experiences with similar systems/controls are incorporated.
Next, the project team can agree on the timing of this initial assessment. For a novel design or site-built system, it may be useful to do this early. It can be done later for established designs. The initial assessment is an engineering tool that may be reassessed if the design changes. It can only be completed when the design is approved (ready to be constructed) to ensure that the final proposed design is assessed.
| Row No. | Operational Sequence | Process Description | SQA/Regulatory Requirement | SPP | Impact on SQA | How SQA Can Be Impacted | Design Controls | Recipe Parameter | Associated Alarm | Procedural Controls | Comments | Residual Risk |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Preloading inspection | Check to ensure unit is visibly clean and dry | N/A | N/A | None | N/A | N/A | N/A | None | The operational procedure will describe how to conduct the preoperational checks | None | N/A |
| 2 | Prerinse | Initial rinse to remove bulk residue and wet dry materials | No loose material, surface wetted | Temperature, fl ow rate/ time (volume/ pressure) | Direct | Particulate not totally removed | The system has low flow rate and high and low temperature alarms; the system will only move to the next step after the process has been completed for the defi ned time with no alarms, trend pressure/fl ow rate data | Yes | Low flow rate and high or low temperature | The operational SOP will describe the action required in the event of an alarm | None | Low |
| 3 | Detergent wash | Initial cleaning stage | Purity (loosening and putting material into suspension/ solution with the cleaning material) | Detergent concentration (conductivity), temperature, fl ow rate/ time (volume/ pressure) | Direct | Variation from the validated cleaning process | The system has low conductivity, low fl ow rate, high and low temperature alarms; the system will only move to the next step after the process has been completed for the defi ned time with no alarms, trend pressure/ fl ow rate data | Yes | Low conductivity, low flow rate, and high or low temperature | The operational SOP will describe the action required in the event of an alarm | Conductivity is used to determine the detergent concentration | Low |
| 4 | Interim rinse | Rinse to remove detergent and removed surface contamination and particulate | Purity (rinsing the wash solution from the unit) | Temperature, fl ow rate/ time (volume/ pressure) | Direct | Remaining surface material contamination of particulate | The system has low conductivity, low fl ow rate, high and low temperature alarms; the system will only move to the next step after the process has been completed for the defi ned time with no alarms, trend pressure/ fl ow rate data | Yes | Low conductivity, low flow rate, and high or low temperature | The operational SOP will describe the action required in the event of an alarm | Conductivity is used to determine the detergent concentration | Low |
The early risk assessment is essentially an engineering tool and may be stored in accordance with project documentation standards. The final assessment of the approved design is a quality document. It should be stored in a qualified document management system along with the associated summary report.
The benefits of this strategy are that a) if high risks are observed, the team has the right expertise to critique it and propose changes to reduce the risks; b) following this approach simplifies a complex process into an easier step-by-step process; c) upon completion of the SRA there is a better understanding of how the system works and the associated risks; and d) there is a list of quality-critical alarms and instruments with the supporting rationale for their categorization.
Facility Fit to Product
Upon completion of the development phase and finalization of the product and process requirements, it is crucial for the project team to review the initially defined generic requirements. This is to ensure that they align with the final requirements. This also includes an assessment of whether the actual CQAs are included in the SQAs and the SPPs include the CPPs. If there is a gap, an update of the SRA with the confirmed CPPs and CQAs added will be required, including any additional qualification required.
The process of verification relies heavily on engineering specifications and is executed by experts in the field. This includes conducting engineering runs and assessing the operational range and precision of equipment, ensuring it can meet future product and process demands. Evaluating equipment suitability often involves two key aspects: confirming that risks to product quality are sufficiently managed and determining equipment performance levels that are acceptable to both the user and the SME.
Should there be any discrepancies between the initial design and the final requirements, adjustments are made under change control and subsequently qualified. This ensures that the system remains compliant and effective.6
Conclusion
In scenarios lacking defined product and process requirements, a generic set of process requirements is used as a design basis, aligning with regulatory expectations. Verification is performed by experts, focusing on equipment performance capabilities and risk to product quality. As per the C&Q Baseline® Guide, equipment qualification covers the operating capabilities of the equipment—these must be assessed against finalized product and process requirements once they are defined to confirm that the equipment is suitably qualified.
In conclusion, the qualification of equipment and utilities in the absence of defined CQAs and CPPs demands a flexible and knowledgeable approach. By leveraging existing data, engaging with experienced SMEs, and focusing on patient safety and product quality, the pharmaceutical industry can successfully navigate the uncertainties associated with the manufacturing of C>s and other drug modalities. This approach not only ensures compliance with regulatory standards but also upholds the commitment to delivering safe and effective therapies to patients.
The qualification of equipment and facilities in the absence of defined CQAs and CPPs demands a flexible and knowledgeable approach.


