Software as a Service: The Journey to Becoming a Life Sciences SAAS Provider
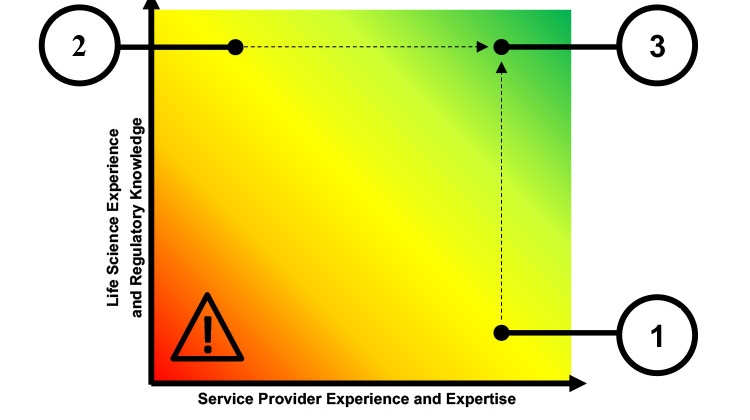
Use of software as a service (SaaS) applications within the life sciences industry is growing. This article reviews the issues and challenges faced by SaaS providers and identifies the qualities of successful providers for this highly regulated industry.
SaaS providers serving the life sciences industry range between two extremes:
- Technology companies that are expert in creating and managing SaaS offerings but lack experience with life sciences regulations (point 1 in Figure 1).
- Mature software vendors with a proven history of supplying applications fit for use with life sciences regulations but with little or no experience with managing a SaaS offering (point 2 in Figure 1).
Companies at each of these extremes will need to adapt and embrace change to ultimately become successful SaaS providers to the life sciences industry (point 3 in Figure 1).
Note that it is not recommended for a regulated company to engage with a provider lacking both SaaS experience and life sciences experience (labeled as exclamation point inside a triangle in Figure 1).
A SaaS deployment model as featured in this article can increase risk for the regulated company, as this model leaves them the least control over the application, its environment, and its configuration.
Many SaaS providers may rely on vendors supplying third-party infrastructure as a service (IaaS) to them. Understanding the associated risks and concerns of such arrangements is important for both the SaaS provider and the regulated company.
“SAAS Smart, Regulation Naive” Providers
SaaS providers that are new to the regulated market, regardless of the maturity and the potential cross-industry applications of their product, may perceive the needs of their regulated customers as invasive in nature; however, failure to address those needs creates a significant barrier for SaaS providers hoping to expand their business into the life sciences. The work SaaS providers do and the artifacts they create in developing, implementing, and maintaining their applications must support their customers’ regulated use of the SaaS system and help them ensure that the system is fit for purpose.
Providers may discover that many of the good business practices already incorporated within their business model may meet the life sciences industry requirements, but it is essential that they become familiar with life sciences terminology so they can explain how their business practices meet the specific regulatory requirements of their customers. Even if the SaaS provider does not use dedicated GxP terms and approaches, they must be able to demonstrate their understanding of the business process their application is intended to support, as well as the use of a defined software development process and controls and operational controls such as security and privacy.
Multiple ISPE GAMP® guidance and regulatory resources are available to help SaaS providers understand life sciences regulations.
ISPE’s GAMP® 5 Guide1 and the supporting Good Practice Guide series provide guidance regarding the basic elements necessary to design, develop, test, and implement a computerized system that is fit for purpose. SaaS providers can use this guidance to understand the expectations and perform a gap analysis between their business practices and regulatory and industry best practices.
In recent years, as smartphones and tablets have become omnipresent, opportunities to create mobile applications that may support regulated activities have increased. The delivery of smartphone applications is almost exclusively the domain of SaaS providers. Guidance on regulatory expectations in this area is available in the ISPE GAMP® Good Practice Guide: A Risk-Based Approach to Mobile Applications.2
Suppliers of software as a medical device (SaMD) need to meet specific regulatory expectations.3, 4 They therefore will have additional demands for their SaaS and cloud providers.
Vendors seeking to provide a SaaS offering in the life sciences industry should pay special attention to ensure that they have the ability to translate and explain their controls and good practices to a customer’s auditor or even (in exceptional circumstances) a regulatory authority. This is discussed further in the section on validation responsibilities.
Regulation Smart, SAAS Naïve” Providers
Driven by regulated customer demand for cloud benefits, many mature life sciences software vendors are transitioning existing products to a SaaS offering, launching new products on a SaaS model, or both.
Often, these vendors are expert in the regulations and requirements for the life sciences industry but may not realize the fundamental paradigm shift in their development and support procedures required to become a SaaS provider.
Paradigm Shift
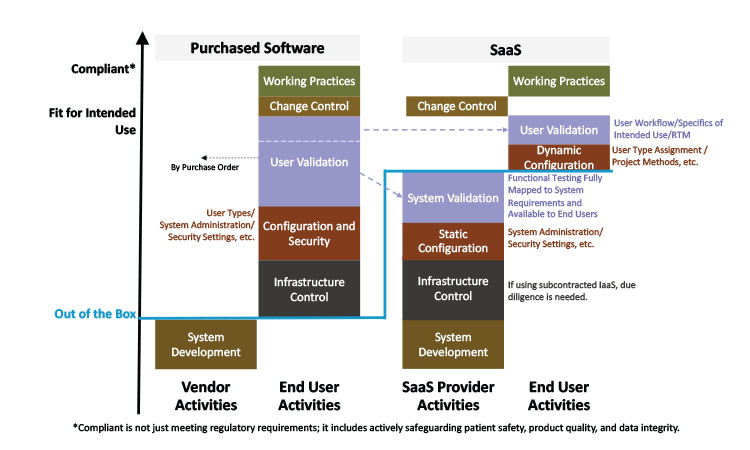
Figure 2 shows the activities of a conventional software vendor and the extent of the additional responsibilities and activities of a SaaS provider. The vendor organization needs to appreciate how much the regulated company’s state of compliance relies on their SaaS provider, and to plan their journey from software vendor to SaaS provider accordingly. As shown in Figure 2, this transition shifts the responsibility and control for many activities from the end user to the vendor. The regulated company will need to rely on many of the activities and the corresponding documentation performed by the vendor to support their activities.
Final responsibility for validation for intended use and compliant operation remains with the regulated company, who are heavily dependent on the scope and quality of activities for system development, infrastructure, configuration, security, validation, maintenance, support, and change control completed by the SaaS provider. The user validation in a purchased software offering is effectively split in two for SaaS:
- System validation—used here to denote the provider’s validation of their SaaS product to demonstrate that system functionality meets their published system requirements.
- User validation—the life sciences company’s validation that the configuration is fit for its intended use leveraging the SaaS provider’s system validation activities.
This topic is discussed further in the section on validation responsibilities later in this article.
System Life Cycle
Software, whether delivered as a purchased product or via a SaaS model, must be developed and managed according to a formal process, with specifications governing the software content, and documented testing verifying that the software meets specifications. The specifics of software development and delivery approaches for SaaS applications are discussed later.
QMS Transparency
Traditionally, mature life sciences software vendors have adopted a robust quality management system (QMS), but they may have limited the information they make available to customers during audits (for example, by restricting access to the internal release testing).
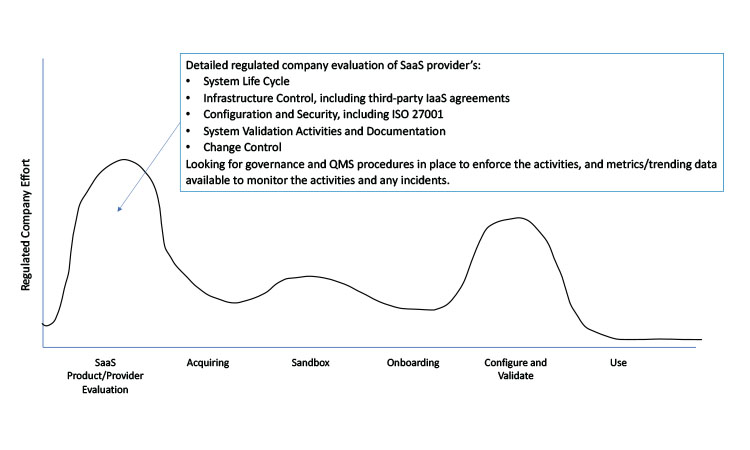
Assessing the SaaS provider’s QMS is a critical and fundamental part of the regulated company’s software selection process. Figure 3 illustrates the relative extent of customer activity from a compliance perspective, from initial assessment to operational use of a SaaS application. Given that the regulated company will depend on the SaaS provider’s infrastructure controls, internal testing, system validation, software release cadence, and so on, it is vital that the regulated company can efficiently and easily assess the provider’s QMS as needed.
Given that the regulated company will become utterly dependent on the SaaS provider’s infrastructure controls, internal testing, system validation, software release cadence, etc., it is vital that the regulated company can efficiently and easily assess the provider’s QMS as needed.
Validation Portal
A validation portal, with access granted only to prospective and current customers, offers a controlled, centralized repository for QMS information, as well as validation information and system documentation, within the bounds of protecting the SaaS provider’s intellectual property. This portal ensures that the regulated company has timely, as-needed access to the latest versions of all of these items, including in support of a regulatory inspection. The validation portal also gives the SaaS provider a place to direct people to change information, risk evaluations, and test scripts.
Keys to SAAS Provider Success
Impact of Provider Choices
When marketing a SaaS product to the regulated life sciences market, the provider must understand the unique delivery and functionality constraints that are critical to successful product commercialization in this market. Successful market penetration, ongoing adoption, and regulatory compliance necessitate that the SaaS provider document, properly architect, and provide a reasonable level of transparency as to the service model they are seeking to deliver. The following sections discuss critical issues that SaaS providers must address to encourage life science companies to accept and use their services.
Infrastructure Controls
Traditionally, the server and network infrastructure supporting life sciences applications was subject to a documented configuration and qualification process. For most regulated companies, the infrastructure qualification process is an accepted part of IT’s job function and has been considered a low-risk activity. However, when organizations migrate infrastructure to the cloud and adopt remotely dispersed, third-party-managed infrastructure, they must perform due diligence to ensure the infrastructure is appropriately managed and monitored by the third-party provider.
There are only a small number of providers who have specialized in offering a traditional life sciences infrastructure qualification approach. Most IaaS providers use automated, real-time management and monitoring systems to ensure the ongoing availability and performance of their service provisions.
As discussed later in greater detail, IaaS and SaaS providers that seek to commercially support the regulated life sciences market should attain and maintain ISO/IEC 270015 certification and/or ensure that SOC 2 Type 26 attestation reports are available. The concepts of risk management, mitigation, change and configuration management, and incident handling can provide regulated life sciences companies with a measure of assurance that the IaaS provider has established ongoing controls that are regularly audited by licensed third-party IT governance professionals. To aid regulated companies’ understanding and acceptance of this approach, several of the larger cloud platform providers have created supplemental materials to trace their internal controls, reports, and third-party certifications to life sciences regulatory expectations.
Tenancy and Data Segregation
As a SaaS provider, there are important distinctions when considering an offering into the life sciences market. If the intention of the application delivery is to provide customers with a fully customized and unique experience, there may be an additional opportunity for the regulated company to control and ensure compliance; however, such applications are unlikely to be supportive of multitenant delivery. Single tenancy is not practically scalable over the long term and would require the addition of resources to manage potentially siloed versions of the customized application.
The additional resources necessary to support single tenancy can place a strain on the service provider, and only a small number of providers offer this. It is more likely that the provider delivers the SaaS application in a multitenant environment. Although the end user has the ability to configure a SaaS application to their intended use, they will, in most cases, lack the ability to heavily customize it.
Multi-tenancy can present a challenge for a regulated company to justify the potential for comingled data on a platform shared by other parties. However, the architecture of modern multitenant database applications is designed to securely segregate each organization’s data, such that these fears are largely unwarranted.
The SaaS service provider should comfortably be able to articulate how data segregation works within the application, and provide transparency around any risk assessment or testing that has occurred to ensure that data segregation and integrity are always maintained.
Certifications and Audit Reports
The pursuit of third-party governance certifications is becoming an acute requirement for a service delivery provider. Many companies will opt to certify against ISO/IEC 27001 or to procure a SOC 2 Type 2 audit report. SOC 2 Type 1 is also popular, but it represents a point-in-time analysis and is therefore less useful to both the service provider as evidence of controlling conformance, and to the regulated company seeking a provider able to prove ongoing good practice. Beyond an 18-month window, the effectiveness of a certification as assurance of ongoing good governance practice may be diminished.
ISO/IEC 27001 certification and SOC Type 2 reports are similar in that they both provide assurance that the SaaS provider has a substantive IT governance process and that this process is in a perpetual state of measurement, monitoring, continual improvement, and risk management. Regulated companies can leverage ISO/IEC 27001 certifications and SOC 2 Type 2 reports7 when seeking to ensure that an IT governance framework both exists and is being managed. Indeed, for a regulated company auditor, many of the concepts, semantics, and outputs of ISO/IEC 27001 would be similar to the QMS they might have in their own IT department.
Attaining the certification and/or audit report can provide immediate assurance to a regulated company that the service provider has a mature understanding of good governance practices and is often leveraged to offset many of the traditional expectations related to installation qualification. Note that a regulated company will also expect that third-party certifications are in place for the SaaS provider’s foundational infrastructure.
Software Development
To comply with the expectations of global regulatory bodies, regulated life sciences companies have, for the best part of the last 25 years, developed and refined approaches for the verification and validation of third-party software applications.
It is vitally important then that SaaS providers that wish to sell their products in the regulated market understand how their system life cycle functions and are prepared to provide a level of auditability that might otherwise be alien to vendors that do not service this vertical market. Providers that are veterans of the regulated software space are generally well informed and understand GAMP® 5 approaches, and are familiar with the acceptable evidence that a regulated company will expect to see. When the software version will be immediately and contemporaneously adopted by all multitenancy users after release, responding using an Agile approach is particularly essential to address any defects or patches quickly. As use cases and user stories are developed, the regulations themselves (21 CFR Part 11, EudraLex Volume 4 Annex 11, etc.) should be held in equal footing as other requirements inputs.
Planning
Most vendors and providers have adopted an Agile approach to software development, which necessitates the creation of sprint planning, and typically a test-first design. Although these are perfectly reasonable approaches and are indeed favored when it comes to the rapid development of high-quality software, the documentation expectations of most regulated companies tend more toward formal, discrete documentation based on past experiences. In turn, the regulated company may need to revise their expectations from using the V-model as a waterfall (linear-sequential) development life cycle, resulting in approved documentation as the output from each stage. Regulated companies will be better served viewing the V-model as a relationship diagram between activities and verifications and the Agile artifacts that record them.
From the outset, the SaaS provider should apply critical thinking to their consideration of what artifacts are required to demonstrate traceability throughout the software development and system life cycle, and how those artifacts can be shared for leveraging by a regulated company. Many software development and/or management tools will inherently include “preservation of information” (artifacts such as specifications, test cases and records, and so on), which may obviate the creation of discrete and formal documentation. A SaaS provider developing and maintaining a high-quality software offering to be sold in the regulated space should ensure that their development and maintenance artifacts demonstrate the traceability of the verification activities against their specifications in whatever tool or format is used.
The planning of a commercial release should consider not only the delivery of functionality but also the delivery outputs needed to satisfy regulated customers. The SaaS vendor must therefore ensure that the tools they use that contain records/data supporting the validated state (e.g., test records) are secure and the records are adequately protected and suitable for leveraging by the regulated company.
Requirements
A working knowledge of the regulations that the SaaS provider’s intended customers must adhere to should be considered as part of the requirements-gathering process when defining the minimum viable product and through all subsequent deliveries. Product managers for software serving the life sciences market should carefully consider regulatory constraints from process and functional perspectives as an additional input source for user stories, use cases, and requirements on the whole. The SaaS provider must also consider which aspects of the final product pose the highest risk to data integrity, patient safety, and product quality.
For example, simply following verbatim the US requirements of 21 CFR part 11 with regard to audit trails and electronic signatures, without understanding the system use and workflows, can result in poorly designed and therefore ineffective or inefficient features and controls.
At the requirements-gathering stage, it is also important for the SaaS provider to consider the infrastructure requirements for the application in scope, the security boundaries, and the tool set to be employed to ensure that data are secure, maintain integrity, and are encrypted as appropriate. Commonly, SaaS-delivered applications have SSL certificates with 256-bit encryption and are monitored by a combination of applications engaged in intrusion detection and prevention, file integrity management, mirroring, and some level of data loss prevention.
Configuration
The definition and management of configuration are best characterized in two ways: static and dynamic. Static configuration is the foundational definition of system settings and interfaces that are necessary to deliver the software in a repeatable and controlled manner. The management of static configuration and any subsequent risk assessment or testing offset configuration should be an integral part of the SaaS provider’s change management approach.
A configuration that is not well managed or is in a persistent state of change can potentially have a downstream effect on the functionality and performance of the application. Though this is true for any SaaS delivery regardless of the target market, the challenges of configuration are more acute in the life sciences market due to the additional layer of regulatory compliance. Changes in the static configuration by the SaaS provider should be assessed for potential impact on the application and verified where needed to confirm the application is not adversely impacted by the changes.
Dynamic configuration involves those items within the application that are configured and managed by the regulated company. Though it would be impractical to test every combination of every potential configuration, the SaaS provider should provide some level of assurance that a typical user configuration consistently meets its intended purpose. Additionally, the SaaS provider can offer customers their insights on suggested configurations and how to test these specific configurations. The provider should ensure that release cycles are planned in advance and well communicated to their customers to allow adequate time for remedial software assurance activities, including user validation.
Risk assessment
SaaS providers often perceive risk through an exclusively commercial lens. Features that could be impactful to the timelines and commercial strategy of the SaaS provider are often prioritized. However, it should be noted that features that are particularly complex to the software architect and engineer may not carry a particularly high risk from a regulatory perspective. It is important to consider an additional facet of risk, namely those aspects of the service delivery or functions of the software that could, given the intended use case of the application, have a potential impact to the integrity of the data owned by the regulated company.
Testing
The software testing process should be holistic, but in the regulated context, the level to which a function is tested is largely driven by its perceived and documented risk to data integrity, product quality, and patient safety. The SaaS provider must be able to provide some level of transparency as to what was tested, the rigor of the testing, and the ongoing change and configuration management plan. These are vital concepts for the SaaS provider to adopt to enable the regulated company to both leverage the testing and gain assurance that the work being done by the service provider can be synergized with compliant workflows at the operational level.
Service providers often do not share information about software testing with third parties because they are concerned about the potential risk to their intellectual property. It would be naive to dismiss that risk out of hand. However, it would be equally naive for the SaaS provider to attempt to sell their product in the regulated space without understanding that customers have a reasonable expectation of a level of transparency about what has been tested, and the process that underlies the testing. This is why the planning aspect from a documentation perspective is an important commercial reality for the service provider.
The level of transparency and portability of the SaaS provider’s testing may correlate with the implementation time, adoption speed, and perceived level of effort for the regulated companies’ own validation requirements.
Software Delivery
The delivery of a SaaS application presents a shared challenge for both the provider and the regulated company. The regulatory and functional aspects of the software should be given as high a priority as any other customer feature because regulated companies will not be able to effectively use the delivered software without these key functional components.
The DevOps concept has fused traditional software development practice and infrastructure management practices into a shared discipline. It is critical then that delivery concepts be considered and weighted just as fully as traditional functional requirements for an as-a-service delivery.
The SaaS provider should look for ways to most effectively deliver their application in a transparent and controlled manner so that prospective regulated customers can have assurance that additional features will not impact legacy functionality.
The SaaS provider should look for ways to most effectively deliver their application in a transparent and controlled manner so that prospective regulated customers can have assurance that additional features will not impact legacy functionality. Providers must also demonstrate through documentation or artifacts how those features were regression tested and verified so that those efforts may be leveraged in the change management process for the regulated company.
The SaaS provider should take the time to provide prospective customers with clear definitions of version changes, patches, emergency changes, and hotfixes. Though there will be semantic differences from company to company, it is important that the provider and the regulated company reach agreement about the processes and process outputs that support these activities.
SaaS providers seeking to serve the life sciences market will find both flexibility and adaptability only within the boundaries of a defined and transparent process—anything short of that will likely be seen as a barrier to acceptance for use by a regulated company, and will therefore be a barrier to the sale.
Validation Responsibilities
As noted previously, life sciences companies expect that the SaaS provider will manage a greater proportion of system validation—the assurance that the system functions correctly according to the provider’s system requirements. The regulated company’s aim is to use the provider’s system validation in support of their end user validation for intended use. However, this does not in any way obviate or reduce the regulated company’s ultimate responsibility for ensuring that any GxP system is fit for their intended use.
Indeed, data integrity guidance from both the US FDA8 and MHRA9 specifically emphasize that validation must be focused on the regulated company’s intended use and cannot be purely a vendor or provider process. EMA guidance10 reinforces a similar concept, stating that the sponsor’s responsibility for the conduct of clinical trials extends to the validation of computerized systems used in support of such trials.
To rely on a vendor’s or provider’s validation and qualification documentation, the sponsor must have knowledge of the vendor’s QMS. This knowledge may be obtained through a vendor qualification process, including an in-depth assessment/audit. The assessment of vendor documentation also helps the sponsor company determine what additional qualification or validation or activities they will need to conduct. Software vendors and SaaS providers should be prepared for these assessments and be able to support the sponsors in meeting their regulatory expectations. Some of the activities often requested by sponsors to assist them in meeting these expectations include:
- Sharing of qualification/validation procedures and documentation during sponsor audit/assessment
- Contractual agreements that vendors will support sponsors during regulatory inspections, including the sharing of qualification/validation documentation and procedures
There is currently an initiative within the US FDA via the Case for Quality program11 to introduce the concept of computer software assurance (CSA). The forthcoming FDA guidance is intended to provide regulated companies with clarity for risk-based software assurance activities. The recent ISPE GAMP® RDI Good Practice Guide: Data Integrity by Design has an appendix tying the CSA concepts to the foundations of GAMP® 5. Key to the CSA concept is a risk-based approach to testing that leverages all previously performed testing.
Because a major principle of CSA is the leveraging of testing from the software providers themselves, adoption of the CSA approach may create a new focal point for both the regulated company and the regulators. It therefore behooves the successful SaaS provider to ensure that their testing, including exploratory, unstructured, ad hoc, and error guessing approaches, affords sufficient rigor, evidence, and traceability that fulfills the regulated customer’s need for system validation. This can, in turn, then be leveraged by the regulated company in support of their user validation and demonstrating fitness for intended use. Robust and traceable provider testing can minimize the impact of software releases and the associated validation burden on the regulated company.
Ongoing Life Cycle
With purchased software, the regulated company has control of what upgrades it implements and when it implements them. In a multitenant SaaS environment, the regulated company is tied to the SaaS provider’s application life cycle throughout the life of their SaaS subscription.
Release cadence
The change management and validation expectations of a regulated life sciences company are often somewhat in conflict with the Agile and perpetual change models that SaaS providers usually prefer. Therefore, the regulated consumer of SaaS software must evaluate whether the application under consideration has a change cycle that presents an acceptable risk profile for its intended use within the regulated company. It is entirely possible that software from an application provider that has an aggressive change cycle coupled with limited documentation is simply not suitable for purchase by a regulated company. When shopping for a cloud-delivered application, regulated companies often look for set change cycles (e.g., semi- or triannually).
A final design review before release for SaaS applications with high GxP impact could provide further assurance to the regulated customer that the final application and delivery checks have been performed.
Support
The support model required for life sciences customers can be similar to the model applied to nonregulated customers. However, SaaS providers wishing to service the life sciences market should have a clear understanding about the level of risk that their applications present to their end customer’s data.
For example, if a manufacturing execution system is cloud-delivered (as many currently are), the loss of data or the delay of transaction processing caused by a service delivery problem could affect the regulated company operations with an immediate and significant financial impact.
Additionally, slow provider response times or overly generous service level agreements (days vs. hours of response time) have the potential to add unforeseen risks to product quality and, in turn, patient safety. It is therefore critical that the SaaS provider align their service level agreement response times commensurate with the risk for the workflows they seek to service. Regulated companies should also be prepared to potentially purchase premium levels of service in order to maintain GxP compliance. The regulated company must also ensure that the SaaS provider’s help desk understands the boundaries of acceptable change during the resolution of a ticket.
CAPA
SaaS providers seeking to support the life sciences industry must understand that although the correction of problems is a necessity, the prevention of their reoccurrence is equally valued. The corrective and preventive action (CAPA) expectations of life sciences companies are aligned with the CAPA provisions of both SOC 2 and ISO 27001. Therefore, attaining one or both of these certifications gives confidence that the SaaS provider uses a CAPA program subject to a regular third-party assessment cycle.
Exit Strategies
The regulated company is ultimately responsible for the integrity of the data supporting their regulated products, and so must consider the full data life cycle.
Extracting and retaining data
The SaaS provider must plan for cancellation of the regulated company’s subscription to the SaaS offering that includes a means to extract the company’s complete regulated data from the SaaS application. Where the data are dynamic, it will be essential that the data can be extracted and passed to the regulated company in a dynamic format. Following the successful extraction, the data should no longer be present in the SaaS application. This may require a documented and authorized manual deletion process.
Readable data for the retention period
Irrespective of the software deployment model or data format, data should not only be retained but also human-readable for a specified retention period, as determined by applicable regulations. When the regulated company has exited the SaaS application, methods to ensure they can read the extracted data must be established. Options include the following:
- If the SaaS provider offers a purchased software version of the application as an alternative to the SaaS subscription offering, the regulated company may be able to install the purchased version on-premise to read the data.
- The data could be converted to be read in an alternative application. However, this approach brings with it the risk of loss of metadata or functionality (especially for dynamic data) and would require data migration verification.
- If there is no other alternative, the regulated company may need to keep a reduced subscription to the application for the purposes of readability of their existing data through their retention period.
Deleting data
Data may need to be deleted from a SaaS application (a) when a regulated company ends their SaaS subscription (after first extracting the data and passing it to the regulated company), (b) when data reach the end of their regulated retention period, and (c) in the case of personally identifiable information, when required to do so under the EU’s General Data Protection Regulation12 or other similar regulations.
Consideration should be given as to whether data will be deleted by the regulated company’s authorized personnel or the SaaS provider’s personnel under written authorization from the regulated company.
Data must be deleted not only from the live instance of the application but also from any archive areas and from backup copies residing in storage. It is unlikely that the regulated company personnel will be able to access and delete backup data; therefore, this task will probably require intervention from the SaaS provider. The SaaS provider may be required to provide an audit trail entry or other record of the deletion to the regulated company as evidence of the deletion.
Conclusion
The life sciences market offers SaaS providers a unique opportunity to positively impact public health and patient outcomes. Many nascent SaaS providers have enjoyed something of a honeymoon in this space, as a desire to adopt their technology has driven many regulated companies to offset inherent compliance weaknesses through internal remediation efforts. As regulated companies’ understanding of “as-a-service” delivery rapidly matures, providers must implement GxP processes or demonstrate that their processes are compatible with GxP to be competitive and maintain legitimacy in the regulated market.
Many of the regulatory requirements for computerized systems and software in the life sciences industry have their foundations in good engineering practices for IT controls. As we have shown throughout this article, the need to meet regulatory expectations should not, and need not, be viewed as an impediment to market. Rather, SaaS providers have an opportunity to increase product quality, ensure data integrity, and apply risk-based approaches and critical thinking to technology applications that fully support customers’ therapeutic areas and patient safety.
About the Authors





