Resilience for Cell Therapy Manufacturers: Five Strategies

For patients who depend on personalized medicine, turnaround time matters. However, moving quickly is difficult for cell therapy companies because designing personalized therapies presents unique challenges unknown in traditional biotechnology. In this article, we’ll examine five strategies to help cell therapy companies develop resilience against these challenges, positioning themselves to reliably deliver what patients need.
Personalized cell therapies use a patient’s own cells (autologous) or donor cells (allogeneic) to treat diseases in ways that traditional off-the-shelf biologics cannot. To deliver on that promise, manufacturers of these novel therapies must navigate a complex, individualized product cycle, starting with cell collection from the patient and moving through processing, cell selection, possible cell modification, expansion, and quality testing steps before finally delivering the finished therapy back to the same patient. Each of these steps must happen quickly, reliably, and without interruption to meet the patient’s need for urgent intervention. The criticality of this manufacturing cycle requires a comprehensive cell therapy manufacturing resilience strategy.
Key Considerations
Manufacturers of all drug types can benefit from improved resilience, but the stakes are elevated in the field of cell therapy. Here’s why:
- The consequences of a lost batch could be life or death. Manufacturers of traditional drug products can safeguard against a product shortfall by building an inventory of contingency stock. However, that is not an option for manufacturers of autologous cell therapies because they must produce batches in real time to meet a specific and urgent patient need. If a batch is underway, that means a critically ill patient needs it. Losing that batch is simply not an option.
- Turnaround time is vital. Reducing vein-to-vein turnaround time has the potential to extend patient lives. One recent study, for example, correlated that cutting the turnaround time of chimeric antigen receptor (CAR) T cell therapies from 54 to 24 days could extend patient life expectancy by 3.2 years.1 Faster delivery may also be a market differentiator for manufacturers, and some may commit to their runtime as part of their regulatory filing. For these reasons, maintaining a predictable turnaround time, safe from unexpected disruptions or delays, is vital.
- Consistency is a challenge. Cell therapy manufacturers rely on living cells as their starting material, derived from a donor or, in the case of autologous cell therapies, from a patient facing a serious illness. Producing a therapeutic vehicle from living cells is a complex challenge. Manufacturers are continuously evolving their approaches and platforms to find potential solutions for that challenge. With few technological solutions on the market capable of handling that development variability, many manufacturers must rely on manual processing steps. This makes it difficult to deliver high-quality and consistent therapies at scale.
- Cell therapies require a different manufacturing philosophy. When expanding large volumes of in-process material, manufacturers of traditional biologics can benefit from the economics of scaling up by using larger bioreactors or production systems to increase batch volumes.2 Cell therapy manufacturers do not have such an advantage. They produce very small batches—sometimes as small as a single patient, in the case of autologous cell therapies. That means they must scale out by adding more production units of the same size to work in parallel.3
Resilience is the key to overcoming these challenges. Resilient manufacturers know where they are vulnerable to delays or downtime, and they have a complex arsenal of strategies for managing that risk. In this article, we’ll look closely at five of those strategies. Among the many interrelated approaches that manufacturers rely on to keep operations running and ensure patients receive needed therapies, these five are especially critical: redundancy, maintenance, automation, segregation, and regulatory compliance.
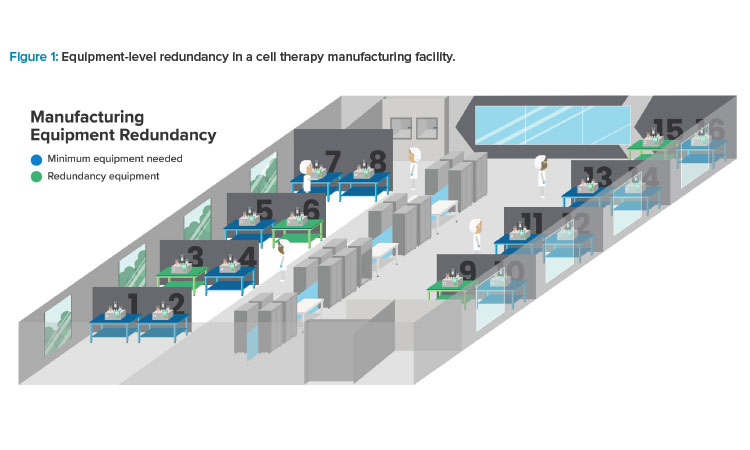
Redundancy
Equipment Redundancy
Owners must weigh the capital and operational costs of installing and maintaining redundant equipment against the business risks of an unexpected interruption.
In a traditional biotech facility
Manufacturing capacity is tied in part to bioreactor productivity. If a production reactor goes offline, that capacity is significantly hindered. Addressing that risk by installing, calibrating, and maintaining an additional “production-ready” bioreactor is an option—but that would mean investing significant capital, time, and footprint in idle equipment. And even if a manufacturer chooses to make that investment, their production system is still vulnerable to resilience issues. What if a system in the seed train goes offline, for example? For these reasons, traditional biotech manufacturers typically build resiliency through other strategies discussed next, rather than focusing on equipment-level redundancy.
In a cell therapy facility
By contrast, equipment redundancy in a cell therapy manufacturing facility is easier to rationalize. The window of opportunity to successfully treat critically ill patients is often very brief; manufacturers are therefore driven to avoid unexpected downtime by investing in redundant equipment. Given the relatively small size of that equipment, installing and maintaining it is easier to justify from a cost perspective than the large-volume systems used in traditional biotech (see Figure 1).
Also, cell therapy equipment is often highly versatile. An automated, closed cell processing unit may support multiple manufacturing steps from cell isolation through transduction and expansion, with programmable options to improve versatility.4 Flexible platforms like this can help cell therapy manufacturers improve the cost effectiveness of their equipment redundancy investments.
It is also important to note that equipment redundancy does not always mean carrying idle systems. Because cell therapy manufacturers must scale out, they have an opportunity to build redundancy into their utilization strategy. For example, a cell therapy manufacturer who requires 12 production systems to meet their throughput target might choose to purchase and install 16 systems, thus building a 25% margin into their utilization rate. All systems will be used, just to a lesser extent. This strategy addresses another important consideration in the cell therapy submarket: cost of goods and its impact on patient access. By balancing equipment failure risk with idle capacity, manufacturers can both establish resilience and manage their capital expenses, which in turn could contribute to better assurances for patients.
Room Redundancy
In any facility, several threats could impact the cleanliness of a production room. A mechanical or electrical failure could cause the room’s air handler or high efficiency particulate air filter to fail. Environmental sensors could detect particles or viables as a result of inadequate cleaning or operational activities. A production spill could both compromise cleanliness and expose in-process materials to cross-contamination. Personnel are widely understood as a significant source of microbial contamination,5 with improper gowning or poorly applied aseptic techniques increasing the risk of environmental monitoring excursions.
Manufacturers can mitigate these risks with rigorous gowning validation, restricted flow of personnel, and continuous monitoring of cleanroom conditions, but the risk that a production room may be compromised remains. To restore it, manufacturers of all types must address the root cause of that event with repairs or remediation, then clean the room appropriately. Depending on the severity of the event, all in-process batches must be either discarded or flagged for quarantine and evaluation by Quality Assurance.
In a traditional biotech facility
Manufacturers carry and manage the risk of a compromised production room. An N+1 strategy in which manufacturers support critical systems with a backup system may offer some protection. However, maintaining entire redundant cleanrooms is not justifiable for the same reasons that rule out equipment redundancy in most cases: the expense outweighs the business risks.
In a cell therapy facility
Cell therapy manufacturers should approach this risk differently, with consideration for the particle concentration permitted for each cleanroom classification, as defined in “Annex 1: Manufacture of Sterile Medicinal Products.”6 For a facility attempting to embrace process closure, the quantity of Grade B suites needed (for open process steps) is often based on how many vectors a manufacturer plans to use or how many Grade C suite activities need support (for closed process steps).
During normal operation, Grade B suites are typically dedicated to a specific product or vector. However, if another suite goes down, a Grade B suite can be leveraged agnostically (following proper cleaning and room turnover protocols). Therefore, manufacturers may benefit from maintaining a single redundant Grade B suite or ensuring that their Grade B suite capacity is sufficient to support normal operations should one such suite be unexpectedly deployed for another purpose.
Grade C suites, where most closed processing activities in a cell therapy manufacturing facility occur, present a different opportunity. Because they operate a large number of small, redundant production systems, manufacturers can put all systems in a single room or distribute them across multiple rooms, depending on their business risk profile (see Figure 2).
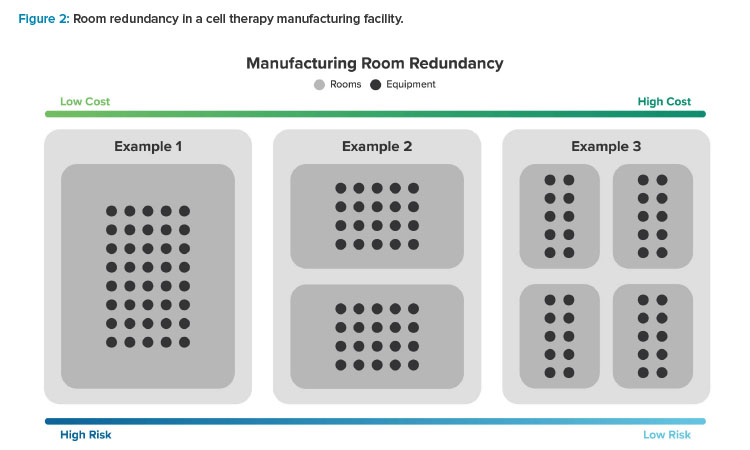
For example, a manufacturer with 40 manufacturing systems may follow the model laid out by traditional biotech facilities, locating all 40 in the same room. Alternatively, they may use two rooms and put 20 systems in each one or use four rooms housing 10 systems each. More rooms mean higher costs—more airlocks, greater facility footprint, more rooms to environmental monitoring (EM) sample, possibly more heating, ventilation, and air conditioning (HVAC) complexity and equipment, etc.—but it also means a lower risk that multiple batches will be compromised by the same room-based contamination event. Manufacturers should consider their business risk profile when weighing these benefits and trade-offs to arrive at a room redundancy strategy that is appropriate for their situation.
Support System and Nonproduction Area Redundancy
In a traditional biotech facility
When access to a facility-based support system is critical, and the costs are justifiable, traditional manufacturers may choose to duplicate that full system. More often, though, they will focus their redundancy strategy on critical subsystems and their component parts. Rather than invest in a redundant water for injection generation system, for example, owners may choose to maintain an inventory of redundant supply pumps.
In a cell therapy facility
For cell therapy manufacturers, this redundancy strategy must go even further. Facility corridors, for example, are vital arteries that keep all processes moving forward. What happens if a contamination event or a failing air handler compromises such a corridor? Without access to the production suites, operators would not be able to perform time-sensitive manual steps on in-process materials. Production would come to a standstill, potentially putting multiple batches at risk. In most scenarios, adding a redundant supply corridor would be impractical. Instead, cell therapy manufacturers should rely on backup air handling units, over-containing protection systems, spill containment protocols, and other contingency measures to proactively eliminate the possibility of a contamination event in the first place.
Power Supply Redundancy
In a traditional biotech facility
To ensure uninterrupted access to power, traditional manufacturers may rely on many layers of redundancy. Sourcing power from separate municipal grids or substations can reduce the risk of an outage; if an outage does occur, battery banks and automated generators can provide short-term backup power to HVAC systems, freezers, and other critical equipment. In addition to these backup systems, traditional manufacturers may also rely on a small uninterruptible power supply (UPS) unit to maintain uptime for their computer control systems.
In a cell therapy facility
Cell therapy manufacturers should approach this risk with a different philosophy. To avoid the possibility of any electrical interruption, many will rely on a larger UPS to support the majority of their manufacturing systems. This impacts the way cell therapy manufacturers design their overall electrical systems. Some invest in photovoltaic panels and other sustainable technologies to generate and store a limited amount of on-site power, with generators on hand to fill the gap. In addition to its decarbonization benefits, this strategy partially untethers manufacturers from vulnerable municipal infrastructure, ensuring facility wide uptime even in the event of an outage.
In a resilient manufacturing facility, processing happens efficiently, unplanned downtime is rare, and fi nal products reach patients rapidly and safely. For cell therapy manufacturers, achieving that vision is imperative: lives depend on it.
Maintenance
In a Traditional Biotech Facility
Resilient facilities of all types rely on preventive maintenance to help identify and eliminate issues before they happen. Traditional manufacturers typically achieve this through planned facility shutdowns, giving maintenance teams a service window of several days or weeks. To de-risk this strategy from a business perspective and prevent shortages in the market during a planned shutdown, manufacturers often draw from inventory stockpiled for that purpose.
In a Cell Therapy Facility
This strategy does not translate well to the cell therapy manufacturing context, where a planned shutdown is more than a business risk—some companies see it as an ethical and moral issue, with patient lives at stake. Personalized medicine cannot be stockpiled, and patients cannot wait for treatment while facility systems undergo maintenance.
The solution is often a strategy of rolling shutdowns, which work hand-in-hand with the redundancy planning described previously. Through careful production planning supported by detailed workflow and schedule mapping, cell therapy manufacturers can pause operations in one area of the facility to perform critical tasks (e.g., Grade B suites require aseptic process simulations twice a year,7 which may identify maintenance interventions required to proactively guard against contamination). Meanwhile, the equipment and room redundancy strategies described earlier ensure that production may continue elsewhere, uninterrupted.
In areas where redundancy is difficult or impossible to achieve (such as in supply corridors), redundant mechanical infrastructure is key—shut down one system for maintenance, keep the other one active for ongoing environmental support. This strategy ensures that all critical infrastructure receives regular preventive maintenance without the need for a full-system shutdown.
However, even the most robust predictive or preventive strategies cannot rule out the possibility of unplanned maintenance. Systems may unexpectedly break, malfunction, or fall out of calibration. To accelerate recovery in such cases, cell therapy manufacturers can rely in part on rented equipment to address specific temporary issues (e.g., compressed gas or liquid nitrogen dewars for malfunctioning utility supply systems). For this strategy to work without causing unexpected delays, manufacturers must have a plan in place to accelerate the qualification process for rented equipment. Preestablished vendor agreements, standardized validation protocols, and preapprovals for certain equipment may play a role.
Manufacturers should also establish a tiered on-site redundancy strategy. Redundant online systems represent the highest tier; these systems are online, in-room, and ready to use. Redundant systems that are stored elsewhere (e.g., in the warehouse) represent the next tier; these systems require installation and some light management but getting them up and running requires less time than repairing the existing system. At the lowest level parts and equipment are stored in the warehouse to facilitate repairs. This tier closely resembles the strategy typically used in large biotech facilities. Cell therapy manufacturers may rely on this tier to support their most expensive and complex systems, such as filling line isolators or autoclaves.
Automation
In a Traditional Biotech Facility
As a pillar of the Pharma 4.0™ evolution,8 automation can enhance manufacturing resilience by reducing operator-driven steps and feeding proactive risk-management strategies with high-quality, real-time data. Traditional biotech facilities meet many of the conditions that make such automation implementations effective and scalable. For one thing, they typically feature standardized processes, making it possible to integrate automation platforms rapidly and effectively. The volume of in-process materials moving through production at any given time also enables an automation-focused manufacturing philosophy. When moving thousands of liters of in-process solution at a time, for example, some degree of process loss during an automated bulk transfer may be acceptable.
In a Cell Therapy Facility
The comparatively small volumes of in-process materials, as well as the manual nature of many cell therapy manufacturing processes, make it difficult for operators in this submarket to leverage the full potential of automated systems at scale. That will change as automation technology evolves—already, the market offers several automated platforms that aim to meet the unique needs of the cell therapy manufacturing life cycle.9 By embracing automated technologies, manufacturers can eliminate manual tasks prone to human variation and replace them with consistent and standardized platforms. This will improve reproducibility and open the door to point-of-care manufacturing, which may translate to lower costs and faster access for patients.10
However, to move into this future of automated cell therapy production, manufacturers must first overcome several challenges. Working with patient cells as a starting material, for example, means accommodating a degree of variability and unpredictable fluctuations,11 which can be difficult to do using automated systems.
Another challenge: many currently available automated technologies are not yet capable of performing the delicate maneuvers required to prevent process holdup loss. Losing even small volumes during a critical transfer (that must be accounted for) could have far-reaching effects on the cost and reproducibility of the final cell therapy product. Estimated material costs for cell therapy manufacturers average about 36% of their overall manufacturing cost of goods, with some manufacturers reporting as high as 70%.12
Material line losses could create a financial impact, which could translate to consequences for patients, whose lives may depend on insurance access to these already expensive therapies.
Although automation is a key to resilient and cost-efficient manufacturing, these challenges have put the widespread adoption of scalable, automated solutions just out of reach for many cell therapy manufacturers—at least for now. New opportunities to bring automation into the cell therapy facility are emerging all the time, thanks to an increasing number of manufacturers in this submarket who are working in partnership with equipment vendors to develop solutions that work for this unique context.13
Segregation
Cleanroom segregation is a critical element of contamination control and operational efficiency. A segregated cleanroom leverages physical and procedural design elements to prevent cross-contamination and ensure that a failure in one processing suite does not impact other production areas, based on a thorough risk assessment.14 This falls within a facility’s contamination control strategy (CCS), which defines the protocols in place to prevent the contamination of sterile or aseptic processes.15 By establishing a robust CCS and supporting it with appropriate segregation, manufacturers can improve their resilience against unexpected downtime or lost throughput.
However, segregation can sometimes work against the goal of operational efficiency. For example, physically separating previral and post-viral processes in monoclonal antibody manufacturing may increase resilience against contamination risks, but this type of room-based segregation will limit efficiencies, drive up both the size of the facility and its cost to operate, and reduce overall manufacturing flexibility. The key is to keep both priorities in balance.
In a Traditional Biotech Facility
Traditional biotech manufacturers have a big advantage when it comes to maintaining that balance: they can leverage process closure technologies to push segregation to the equipment level rather than relying on room classifications and environmental segregation to prevent contamination. This can open the door to a simpler, more scalable, and more cost-effective facility layout, such as a ballroom design in which multiple processes are completed in parallel, with mobile equipment that can be reconfigured as needed.16
In a Cell Therapy Facility
Cell therapy manufacturers should think about this calculation differently. As established in the preceding section, full end-to-end process automation at the equipment level can be difficult to implement. As a result, many manufacturers are tethered to a room-level segregation strategy, depending on the degree of automation and process closure they are able to leverage. From an efficiency perspective, this can make it difficult to optimize the cell therapy facility.
The complexity of scaling out rather than up adds to this challenge. As discussed in the section about room redundancy, processing potentially hundreds of small batches in a single shared cleanroom would generate a high degree of efficiency. However, a single HVAC issue could compromise every batch. Distributing these batches across multiple rooms lowers that risk but may increase the facility’s footprint and drive up both capital and operational expenses, considering that every additional room would require manufacturers to replicate and scale out all room-based functionalities (e.g., airlocks, in-process testing stations, staffing plans, such as supervisors, EM sample locations, and so on).
Given these choices, how can cell therapy manufacturers balance segregation and efficiency to support ongoing resilience? The answer will involve some degree of risk-based redundancy, calculated function by function, to ensure continuity of operations in the event of a contamination event. Broadly speaking, manufacturers should consider at least some degree of segregation at the room level, depending on how many batches product types they intend to manufacture concurrently.
Regulatory Compliance
For any manufacturer of a drug product carrying a sterile claim, supporting that claim with appropriate facility and process designs is a critical priority. It is the key to ensuring patient safety and maintaining compliance with relevant guidelines.
In a Traditional Biotech Facility
Traditional biotech manufacturers typically apply a sterile filtration step to remove microorganisms and particulates from final product formulations. Downstream of the sterile filter, they must comply with “Annex 1: Manufacture of Sterile Medicinal Products”17 and the “Guide to GMP for Medicinal Products.”18 Although Annex is not formally adopted by the US Food and Drug Administration (FDA), US-based manufacturers generally choose to follow the guideline’s principles to ensure patient safety and compete on a global scale.
Upstream of the sterile boundary, manufacturers comply with “Annex 2: Manufacture of Biological Active Substances and Medicinal Products for Human Use”19 aiming to control bulk drug bioburden levels. Efficient measures like process closure, robust cleaning protocols, and ongoing monitoring and validation help maintain low microbial loads as bulk drugs move toward sterile filtration and downstream aseptic processing.
In a Cell Therapy Facility
Compliance is more complicated within the context of cell therapy. In 2017, the European Commission published EudraLex Volume 4, Part IV “Guidelines on Good Manufacturing Practice Specific to Advanced Therapy Medicinal Products.”20 Four years later, the Pharmaceutical Inspection Co-Operation Scheme revised its “Guide to Good Manufacturing Practice for Medicinal Products” annexes to include Annex 2A, “Manufacture of Advanced Therapy Medicinal Products for Human Use.”18
Regulatory updates provide cell therapy manufacturers with a set of GMP guidelines dedicated to their manufacturing context. Although Annex 1 is not strictly mandated by Part IV, many cell therapy manufacturers aim to align with Annex 1 principles. Achieving alignment can be challenging in cell therapy. Sterile filtration is not possible for the process stream, given that pore size in sterilizing-grade filters is 0.2/0.22 μm21 and typical human cells are 25 μm. 22 As a result, the whole manufacturing cycle might have to be performed aseptically. To achieve this, cell therapy manufacturers must rely on their evidence-based quality risk-management program to identify and redress contamination vulnerabilities at any point in production.23
A CCS can help cell therapy manufacturers achieve this aim.15 Although it is not required, US-based cell therapy manufacturers may use a CCS as a signal to regulators of their robust commitment to identifying and addressing potential contamination risks across the cell therapy life cycle. The CCS encompasses many of the elements discussed throughout this article, as well as components such as cleanroom classification, cleaning regimes, pressurization schemes, gowning practices, and more. It is fundamental to establishing a resilient cell therapy manufacturing strategy that protects critical in-process solutions from the possibility of contamination.
Conclusion
In a resilient manufacturing facility, processing happens efficiently, unplanned downtime is rare, and final products reach patients rapidly and safely. For cell therapy manufacturers, achieving that vision is imperative: lives depend on it. But it is also uniquely difficult. To get there, cell therapy manufacturers must learn and diverge from traditional biotechnology manufacturers, ultimately building a strategy for resilience that centers on the patient and their time-sensitive need for high-quality, life-saving personalized therapies.


