

A Beginner’s Guide to IT System Inspection Readiness

This article provides a beginner’s overview of how organizations can achieve a state of preparedness (readiness) for inspections, with a specific focus on IT systems.
Computerized systems are increasingly integrated into the pharmaceutical business, including within regulated Good Laboratory Practice (GLP), Good Clinical Practice (GCP), Good Pharmacovigilance Practice (GPvP), and Good Manufacturing Practice (GMP) domains and supporting activities. In turn, regulatory authorities conduct inspections to ensure that life sciences companies, clinical research teams, and other related organizations understand and comply with the regulations established to protect patient safety, product quality, and data integrity. As a result, computerized systems are a key focus area during audits and inspections.
“Computerized systems” is a broad term. In this article, it will be a synonym for process control systems, laboratory equipment with embedded computerized systems, and applications used for GxP purposes in general that are made up of infrastructure, software, supported processes, and “people aspects” such as training and qualification. This definition can relate to solutions from cloud-based services such as infrastructure, platform, and software as a service (IaaS, PaaS, and SaaS) or on-premises solutions.
Resources
Simple questions often have extremely complex answers. It may take time to find the right individuals to provide the appropriate information.
Information on general approaches to inspections of computerized systems and the documentation to support such inspections can be found in:
- ISPE GAMP® 5 Guide: Compliant GxP Computerized Systems1
- Appendix M6: Inspection Readiness in ISPE GAMP® Guide: Records and Data Integrity2
- US FDA’s “Inspections of Computerized Systems in Drug Processing”3
- Section 24 (checklists and memory aids) in the Pharmaceutical Inspection Convention/Pharmaceutical Inspection Co-Operation Scheme’s “PIC/S Good Practices for Computerised Systems in Regulated ‘GxP’ Environments”4
General requirements to support inspections of computerized systems can be found in:
- Chapter 7: Outsourced Activities in “EudraLex, Volume 4: EU Guidelines for GMPs for Medicinal Products for Human and Veterinary Use”5
- Annex 11: Computerised Systems in “EudraLex, Volume 4”6
- US FDA Compliance Policy Guide, Section 425.200, Computerized Drug Processing; Vendor Responsibility (CPG 7132a.12)7
- EMA’s “Q&A: Good Clinical Practice (GCP)”8
Preparing for Inspection
Regulators usually announce inspections ahead of time. Depending on the type of inspection, they may commonly provide between two and six weeks’ notice.
Once notice is given, the initial activities are to identify and confirm the scope of the inspection, its location, and the type of inspection (e.g., new drug application [NDA], routine, or a combination thereof). This information will help to identify which IT systems will potentially be covered within the scope and remit of the inspection.
An organization should plan to be inspection-ready at all times, and to support this, it is necessary to have a governance structure led by an inspection steering committee that can coordinate several key activities, including the following:
- Providing an overview of IT systems in scope and the related business processes
- Managing communication setup, planning, and availability of persons to be involved (e.g., system owners, system managers, specialists, and suppliers)
- Handling any necessary gap analysis activities, including corrective actions (refer to the checklist mentioned later in this article)
- Preparing technical setup for inspections (e.g., demonstration of the IT system, tours of facilities including data centers)
- Ensuring it will be possible to respond to technical questions and provide evidence at short notice (e.g., test evidence related to backup and restore processes)
- Planning mock inspections if the organization is not accustomed to receiving regulatory inspections
- Planning the process for provision of electronic records in a secure format to the regulator if required during, or after, the inspection
It is the steering committee’s responsibility to ensure the organization is well prepared for upcoming inspections. A good way to substantiate this preparedness is to hold mock inspections so that everyone involved becomes familiar with inspection procedures and expectations of the process.
Organizing during Inspection
During the conduct of the inspection, several activities will require attention. They can be briefly summarized as:
- Collecting, interpreting, distributing, and controlling questions raised by the inspector
- Locating the appropriate documentation and delivering this to the inspector upon request in a timely manner
- Coordinating answers and documentation to the inspector
- Communicating daily with relevant parties to share information on questions raised, answers, and the plan for the next day of the inspection
Inspection and Preparation Rooms
For practical purposes, it is valuable to have at least two dedicated meeting rooms established for the inspection process: one room designated the “inspection room,” where the inspector conducts requests plus a room (or two) where the organization’s employees may prepare.
The inspection room is where the inspection is hosted and the dialogue between the inspector and relevant employees is conducted. It is recommended to assign an employee to the role of secretary to record requests. For the secretary, the use of an online/real-time tool is useful so individuals supporting the inspection within or outside the inspection room can see the requests. It is vital that those in the preparation room(s) have the ability to follow the progress of the inspection in the inspection room without interrupting the inspector.
During the inspection, one or more support staff members should be assigned to serve as runners. Their responsibilities are to move between the inspection and preparation rooms, coordinating the requests and provision of responses, while also escorting additional people, as required, into the inspection room to respond.
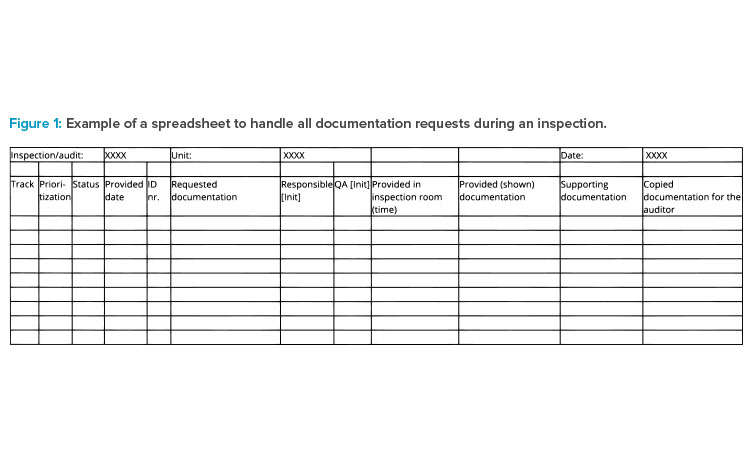
All coordination for the inspection takes place in the preparation room(s). In this space, employees manage all requests for materials and plan and prepare those materials for presentation to the inspector. Runners communicate timelines and status from the preparation room(s) to the host (typically an accountable person from the business and quality management team) in the inspection room. Figure 1 demonstrates how documentation requested for the inspection can be tracked.
Members from the steering committee or management team related to IT systems are represented in the preparation room(s), where they continuously follow the trend of questions and identify potential IT areas/systems within the inspection scope. Based on this surveillance, the prioritization of activities at the system owner/manager level can be determined and communicated. Particular lines of questioning associated with an IT system (for example, access control, backup testing, or data integrity) can also be communicated to those responsible for other in-scope systems during the inspection to allow them to prepare for similar questions and provide responses in a timely manner.
The use of preparation rooms also allows coordination of the flow of people into the inspection room to present materials and respond to questions. Depending on the size of the team and the office setup, a separate room could be used for this purpose. The goal is to limit the number of people within the inspection room at any one time, and to ensure that the movement of people in and out of the room does not disturb the flow of question-ing/topics from the inspector’s perspective.
It is important that a person experienced with inspections oversees the person answering the inspector’s questions and providing supporting documentation. To further support the inspection in the best way, the person answering the inspector’s questions is typically expected to:
- Provide a brief description of the working procedures and documents
- Answer questions from the inspector
- Seek clarification for any questions that are not clearly defined or understood
During preparation, it is recommended that attention is focused on the following areas to prevent misunderstandings or errors:
- Ensuring that the documentation presented answers the questions asked
- Ensuring that the result/conclusion is clear from the documentation presented
- Reviewing the documentation to make sure that it can be used: for example, no notes pinpointing faults are attached, all applicable signatures are applied, and the correct document version is presented
- Applying tags (#1, #2, etc.) to longer documents, as appropriate, so the relevant information can be easily located
- Presenting only the information necessary to support the answers to questions raised, while excluding too much or irrelevant documentation
- Ensuring that all documentation is copied, registered, and stamped in line with the organization’s documentation policies
When these tasks are satisfactorily completed, the person is “released” as ready to enter the inspection room. Staff in the preparation room(s) will manage this entrance; it is recommended that the person attends the session before their allocated time slot to help them become comfortable with the style of the inspection and the atmosphere in the room.
Case Example
The following is an example of IT systems–related questions provided by an EMA inspector on a GxP application about the organization’s handling adverse events, and the related preparation activities.
- The inspector’s questions are:
- How do you manage backup?
- Is the backup qualified?
- I would like to see the disaster and recovery plans.
Preparation Room Discussion
To address question 1 (How do you manage backup?), staff in the preparation room cover the following issues:
- Are procedures in place internally and at the service provider as well?
- Are all servers included in the backup according to the configuration item list for the application?
- If the backup has failed, do we follow the procedure? How is information provided between the service provider and us?
- What backup method is used?
- What is stated in the agreements (service level agreement/statement of work) between our company and the service provider handling backups?
For the inspector’s second question (Is the backup qualified?), key areas for preparation are:
- What version of
is used? - Are all the required documents in place (qualification)?
- What has been included in the challenge test supported by the conducted risk assessment?
- What is the split of responsibilities between the backup service provider and the system/application owner?
- Are there any issues with the backup system which we need to be aware of?
For the inspector’s third request, about the disaster and recovery plans, the staff in the preparation room review:
- Are procedures in place specifically for the applications?
- What is the split between our procedures and service provider procedures?
- When was disaster and recovery planning last tested?
- What is the process for data recovery?
Inspection Room Strategies
The employees meeting with the inspector provide a high-level explanation that
- Having documentation about daily backup operation procedures ready to show to the inspector
- Providing the name of the backup system
- Explaining that the servers are backed up daily following the standard operating procedure (SOP), which also describes the retention period, and being prepared to present the SOP documents
- Confirming that the backup system is qualified according to the quality management system (QMS) process, and having any relevant reports available to present
- Confirming that backup and restore functionality is a part of qualification
Regarding the inspector’s request to see the organization’s disaster and recovery plans, the employees explain that the overall disaster and recovery plans for the service provider and central backup system are confidential and may not be disclosed to third parties (a reference to the specific company policy is provided). The employees further explain that the overall plan is implemented by an instruction in both the service provider’s QMS and the company’s own QMS, and they have the instruction available to show to the inspector.
Finally, to demonstrate that the organization properly conducts backups, the employees are prepared to provide evidence for the previous week’s completed backup.
The Checklist
Based on this author’s experience, the regulated company should always be able to answer five basic questions about computerized systems:
- What does the system do?
- What are the potential product quality, patient safety, and data integrity aspects of the system?
- Who has access to the system?
- What does the system consist of?
- How do you know the system works as it should/is it fit for intended use?
The checklist in the Appendix (available at ISPE.org/it-systems-checklist) can be used to prepare to answer these questions. It is built upon good practice gained from knowledge of auditing, previous inspections, and guidelines from the authorities. It does not cover all situations. However, if the company can respond “yes” to all questions and individuals are able to practice answers and responses, significant problems are unlikely to occur during the inspection. The checklist is generic and therefore does not take into consideration any local requirements.
Specific Scenarios
The following sections review some scenarios that can be especially challenging during inspection of computerized systems.
Providing an Up-to-Date Inventory
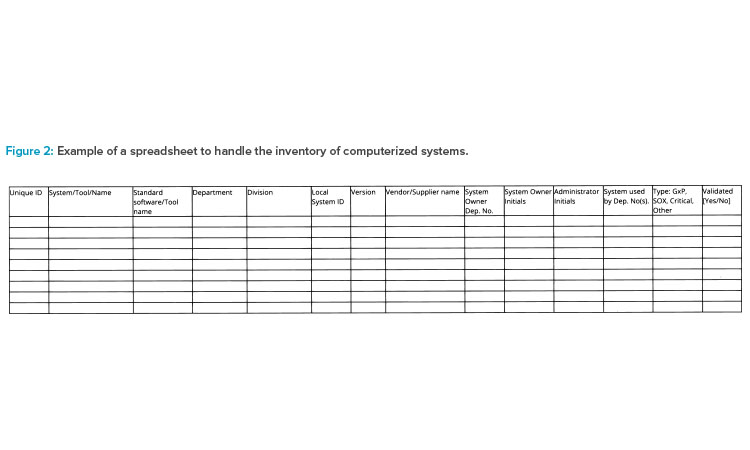
Larger companies should have a dependable enterprise architecture in place to provide an up-to-date inventory of computerized systems when inspected. Some companies have a holistic IT solution that covers all information; other companies have a local inventory list in each department and then consolidate those lists as a part of the inspection-readiness process. Figure 2 illustrates a spreadsheet tool for inventorying computerized systems.
The following data could be stored in a central repository, or system inventory, to support inspections:
- Software/service name and possible vendor(s)
- Software category
- Current version
- Original go-live year
- Criticality
- Life-cycle status
- Level of user access
- Security measures
- Link to user guides
- Agreements/operational level agreements
- System access
- Dependencies’ business processes
- Responsibilities
- Interfaces
The process of maintaining the system inventory should be described in an SOP. If those data are not held in a central repository such as a system inventory, a job aid specifying the location and points of contact could be useful.
Inspector Access to Computerized Systems
In the past, it would have been very unusual for an inspector to request direct access to systems. When such requests are made, the host will usually have an experienced system user log on to the system and then the inspector indicates which transactions or data they wish to see. Demonstration of systems should be planned in advance to make sure relevant functionality and related data are shown appropriately.
A significant barrier to inspector access is the need to create an approved user ID and provide the appropriate system training to the inspector. Typically, inspectors should only be granted access to a guest network for administrative purposes. During an inspection, segregation between the internal network and guest network must be possible.
Requests for Electronic Computer Records
During inspections, it is not unusual for an inspector to request an electronic copy of records from the computerized system. Inspectors may also request access to audit trail data, which they then analyze using tools that search for unusual patterns in the audit trail that could hint toward data integrity issues.
When considering such requests, the company should clearly understand what data are requested, in which format records should be provided, and on what media. When providing an electronic copy of a record, it is preferable to use a validated export function in the computerized system. If data are to be extracted from a database or similar system, it might be necessary to make a script to transfer the data. This script must be specified, reviewed, and tested to make sure it works properly. In all cases, the method for copying electronic data should be documented.
To verify what data are delivered to the inspector, a digital fingerprint on the files should be made. This could be done via an MD5 (message digest algorithm 5) hash code. This is documented together with an electronic copy of the record. In addition, company security protocols (e.g., regarding the use of removable media, virus checking, and data encryption) must also be followed.
Global Multisite System Inspection
Some systems (e.g., enterprise resource planning) might be maintained at the corporate level. If the inspection is at a site level, it is important that the system owner can provide the necessary support to the local site. In general, validation of generic features is done centrally, and specific flows and functions are validated locally. This split should be clearly identified in the validation documentation and associated SOPs. As a part of the planning for inspection, planning participants should determine who will provide what kind of evidence to inspectors. Special attention should be given to SOPs covering how change control for the IT system, risk management, and incidents are managed between the corporate level and site level.
Supplier Involvement
In today’s world of cloud-based systems (IaaS, PaaS, and SaaS), much of the essential documentation discussed in this article may only be available from the cloud computing service provider (supplier).
The QMS at the regulated company should include the management of outsourced activities because the regulated company retains ultimate responsibility for the system validation and therefore must ensure processes are in place to ensure the control and review of outsourced activities. The regulated company is responsible for the suitability of computer systems (hardware and software) used in the manufacture, processing, or holding of a drug product.
Basic Recommendations
As a minimum, it is recommended that organizations ensure the following:
- Quality system and audit information relating to suppliers or developers of software and implemented systems/services is made available to inspectors on request.
- The organization has formal quality agreements with all suppliers, and these agreements include clear statements to:
- Define the system owner’s and suppliers’ roles in making and maintaining original documents or true copies in accordance with cGMP or other GxP regulations
- Explain how those records will be made readily available for inspection
- Indicate that electronic records will be stored in accordance with cGMP or other GxP regulations and will be immediately retrievable during the required record-keeping time frames established in applicable regulations
- Indicate how suppliers will support inspections both through specialists and documentation
- The organization documents its monitoring and review of the supplier’s performance, including the identification and implementation of any required corrective and preventive actions or improvement(s).
Requirements for Clinical Trial Sponsors
Regarding the level of qualification/validation required of a clinical trial sponsor when using an electronic system previously qualified by a supplier, the EMA Q&A for GCP states:8
The system in question may be a system validated by the supplier, but installed at the sponsor, or a system provided as software-as-a-service (SaaS or cloud solution).…
According to ICH E6(R2), sections 5.2.1 and 5.5.3.a, respectively, “the ultimate responsibility for the quality and integrity of the trial data always resides with the sponsor” and “the sponsor should ensure and document that the electronic data processing system(s) conforms to the sponsors established requirements for completeness, accuracy, reliability, and consistent intended performance (i.e., validation).”
According to ICH E6(R2), section 1.65., validation of computerized systems is “a process of establishing and documenting that the specified requirements of a computerized system can be consistently fulfilled from design until decommissioning of the system or transition to a new system.”
The sponsor may rely on qualification documentation provided by the supplier if the qualification activities performed by the supplier have been assessed as adequate. However, the sponsor may also have to perform additional qualification/validation activities based on a documented risk assessment.
The conditions for a sponsor to use the supplier’s qualification documentation include, but are not limited to, the following:8
- The sponsor has thorough knowledge of the vendor’s quality system and qualification activities, which will usually be obtained through an in-depth assessment/audit.
- An assessment/audit has been performed by qualified staff, with sufficient time spent on the activities and with cooperation from the vendor.
- An assessment/audit has gone sufficiently deep into the activities, and a suitable number of examples for relevant activities has been reviewed (and documented).
- The assessment/audit report determined that the vendor’s qualification documentation is satisfactory, or that shortcomings can be mitigated by the sponsor (e.g., the sponsor is performing part of the qualification).
- When required during a GCP inspection, the qualification documentation is made available to the inspectors in a timely manner, irrespective of whether it is provided by the sponsor, the contract research organization, or the vendor.
- Both the sponsor and the vendor establish full configuration management for qualification and production environments, and establish that the sponsor can fully account for any differences between the vendor’s validation environment and the sponsor’s production environment; subsequently, the sponsor should justify any differences that are considered insignificant. If this is not done, the qualification effort potentially does not justify the use of the system.
- The sponsor performed an installation qualification/performance qualification if the system depends on trained users.
The EMA Q&A for GCP notes the following about potential pitfalls in contractual relationships with electronic systems vendors in relation to clinical trials:8
Special consideration should be given on relevant training and quality systems. Experience suggests that vendors accepting tasks regarding electronic systems are frequently knowledgeable about IT systems and sometimes data protection legislation, but not necessarily on ICH E6(R2) requirements, quality systems, etc.
Vendors must be able to document knowledge on, for example, GCP and compliance. Agreements should state that the clinical trial sponsor can access conduct audits at the vendor site, and that the vendor site agrees to allow inspections by national and international authorities. In addition, agreements need to specify that vendors shall provide the necessary documentation (e.g., qualification documentation prepared by the vendor concerning the system) when requested during a GCP audit/inspection process.
Vendors should have appropriate documentation in place. Any qualification documentation prepared by the vendor concerning the system should be available for inspection.
Furthermore, agreements should stipulate that the vendor will escalate any potential serious breaches to the sponsor in a timely manner. Serious breaches should be defined to include security breaches which the supplier becomes aware of (e.g., by notification from other sponsors using the same system), if the breaches could have any impact on the data integrity, reliability, and robustness, or the safety and rights of the trial subjects.
Data Integrity
Data integrity is a topic that spans processes and products in the regulated company. Some of the recommendations already mentioned in this article refer to data integrity. In addition, special attention should be given to the following areas.
General Procedures
General procedures focused on data integrity must be in place. Regulated companies should:
- Consider record and data integrity within the context of broader inspection-readiness programs
- Establish and maintain policies and procedures that ensure a constant state of inspection readiness
- Have robust established procedures for all aspects of the system life cycle
- Be prepared for regulatory inspections:
- Focusing on the management of record and data integrity to verify the adequacy of controls
- Using a forensic approach that challenges the data integrity of specific records
Accountability
The process owner and system owner are normally accountable for responding to system-specific questions during regulatory inspections. Process own-ers and system owners should be:
- Knowledgeable about the documentation supporting the implementation, control, maintenance, use, and history of the system
- Able to discuss any technical and procedural controls implemented to support the integrity of the creation, processing, and reporting of records and data
- Able to share information about the requirements and testing of the data integrity relating to technical and procedural controls
- Able to discuss the key computer system documents including requirements for data integrity controls and system security controls
Procedure Monitoring
From an operational point of view, there should be robust monitoring of the system, business, and IT support procedures to ensure that the processes are adequate and are being followed. Areas that should be routinely reviewed as part of monitoring to ensure inspection-readiness include:
- Access control:
- Access SOPs are in place and being followed.
- Available user roles are documented and managed by change control.
- Documentation shows that only authorized and trained people have system access.
- Evidence shows that access is periodically reviewed (by automated checks where available).
- Segregation of duties is enforced.
- Generic accounts are not used for data modification.
- Backdoor changes requiring IT tools and skills are authorized, verified, and documented.
- Historic access records are properly retained.
- Backup and disaster recovery:
- Procedures for backup, restoration, disaster recovery, and record retention are documented and verified.
- Documented evidence shows that records and data are periodically backed up.
- Records retention policies are clearly defined and followed.
- Records and data can only be accessed by authorized users (network and system).
- Archived records are secure and accessible for the retention period.
- Record and data maintenance is done correctly.
Audit trail:
- Use SOP governs the timely recording of data.
- Records are approved/signed only by authorized users.
- Approvals are enforced at specific points in the business process.
- Audit trail review (in accordance with risk) is integrated into the business process.
Conclusion
The purpose of inspections is to demonstrate to regulators that the regulated company complies with requirements and implements controls in their QMS with the goals of patient safety, product quality, and data integrity. Inspections are more likely to have a successful outcome when organizations follow recommendations for validating and maintaining computerized systems. Further recommendations and guidance to support planning and preparing for inspections are available from ISPE.1, 2
Above all, remember the 5Ps: Proper planning prevents poor performance.
About the Author



