2022 ISPE Europe Annual Conference Madrid: In-Person Again

The ninth ISPE Europe Annual Conference was the first in-person conference after a break of over two years due to the pandemic. For travel-related reasons, some attendees participated remotely. With 450 participants on-site and nearly 490 total attendees, the usual atmosphere of an ISPE conference was present.
The keynotes addressed biologics—the fastest-growing sector of pharmaceutical drugs—and among those, cell and gene therapy approaches as well as personalized medicines. Biologics are a challenge for all functions in operations departments: from research and development to production, engineering, and IT to quality and supply chain excellence. Including cross-functional teams is a success factor for the future of biologics.
Representatives from the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) discussed the role of regional regulatory agencies in addressing current challenges for the pharmaceutical industry and suppliers, and public health and safety. Top priority is given to avoid drug shortages; improve accessibility to vaccines; further innovation; and driving digital transformation, sustainability, and continuous improvement of quality and GXP compliance as the essential ground of patient safety.
ISPE’s Facility of the Year (FOYA) category winners were presented, with their lighthouse projects to disclose the future. More detailed information about specific sessions follows.
AEMPS Overview
Maria Jesus Lamas Diaz, Chair of Agencia Española de Medica-mentos y Productos Sanitarios (AEMPS), the Spanish health authority, opened the Executive Forum with an overview of AEMPS’ national and international activities, as well as Diaz’s personal involvement in European committees. The initial impact of COVID-19 was supply chain disruptions, hindered production, and a sudden increase in demand.
From AEMPS’ perspective, the ongoing supply chain needs are continuous monitoring of sales, existing stocks, and scheduled new batches. AEMPS also anticipates peaks in demand and control of supply for certain products. The continuity plan put forth by the regulatory authority has the following elements: (a) GXP inspections completed remotely as long-distance assessments due to travel restrictions; (b) the need to support marketing authorization and variations procedures, and to implement regulatory flexibility policy, agreed upon at the European Union (EU) level; (c) the need to tackle shortage problems; and (d) enhanced support for COVID-19 vaccine development (e.g., by providing GMP inspection resources).
Digital Transformation
Marco Odoardi, Senior Director, Head of Global Warehousing and Distribution for Merck KGA, continued the conference with a session focused on digital transformation of the pharmaceutical industry and the Pharma 4.0™ holistic approach within Merck Healthcare.
Digital transformation allows the pharma industry to shape the future and to gain and sustain competitiveness in time. “Digital transformation starts as a trickle…and builds to a flood,” said Odoardi. It allows organizations to fully live up to their accountability and to operate with autonomy and command of the products. It goes hand in hand with the increasing digital maturity, being closer to the business to accelerate value creation, and being a competitive advantage to the company.
Odoardi discussed the structured Merck approach, which is built around three dimensions: Set the aspiration, deliver higher-priority use cases, and sustain it. “A particular emphasis will be placed on the culture and people involved,” he said, noting that business drivers and logic are “sterile” if the required mindset and behaviors are not embedded in the organization.
Merck targets “where people are the success factor and will be game changers in the external environment on the successful digital transformation.” There is no magic bullet for successful digital transformation, he said, and it won’t happen overnight. But there are best practices and ways to create conditions for success. Fostering a digital-impact-driven culture and driving people development are key to ensure replicable success.
Technology Trends
Paul W. Rutten, Partner, McKinsey & Co., led a session entitled “Tech Trends that Matter.” Advanced technology has always spurred economic development, and now it’s accelerating even faster, he said. In the next decade, the world will experience more progress than in the past 100 years combined as technology reshapes health and material sciences, energy, transportation, and a wide range of other industries and domains. This progress will have broad implications for the industry.
Each industrial revolution was driven by advanced technology—the first by the steam engine (1769), the second by the internal combustion engine (1867), and the third by the internet (1970s). The fourth will be driven a range of scientific research streams collectively known as “omics,” Rutten said. McKinsey plans to research technologies that will fuel the fourth industrial revolution as well as the impact from the upcoming bio revolution.
Omics include three main categories: intracellular flow of genetic information, intracellular products of metabolism, and others. The first, intracellular flow of genetic information, includes epigenomics, epigenomic DNA modifications, transcriptomics, and proteomics, which are described next:
- Epigenomics: The full genetic complement of an organism; relatively static over time.
- Epigenomic DNA modifications: Epigenetic marks that regulate gene expression (e.g., DNA methylation, histone protein modification).
- Transcriptomics: The complete set and quantity of RNA transcripts that are produced at a given time.
- Proteomics: The entire set of proteins of an organism with changes over time.
The second, intracellular products of metabolism, includes metabolomics, glycomics, and lipidomics, which are described next:
- Metabolomics: A set of metabolites, small molecule intermediates, and products of metabolism.
- Glycomics: The structure and function of the complete set of glycosylated products (e.g., glycans).
- Lipidomics: The complete set of lipids produced.
The third category describes other omics: microbiomics, single-cell omics, and circulating cell-free DNA or RNA analysis:
- Microbiomics–Microbe population: All microbes in a population (e.g., the human gut).
- Single-cell omics: Includes human and other cells and captures single-cell-level nuances that aggregation across multiple cells would miss.
- Circulating cell-free DNA or RNA analysis: DNA/RNA in bloodstream, not in cell; noninvasive or transcriptome information.
Digital Therapeutics
Giuseppe Recchia, CEO, daVinci Digital Therapeutics, addressed the barriers and challenges within digital therapeutics. One of the main issues in this sector is missing or incomplete definitions of terms. For example, digital health includes technologies, platforms, and systems that engage consumers for lifestyle, wellness, and health-related purposes to capture scores, transmit health data, and potentially support life science and clinical operations. However, programs within digital health do not require clinical evidence or regulatory oversight.
Digital medicine includes evidence-based software and hardware products that measure or intervene in the service of human health and that require clinical evidence and regulatory oversight. Digital therapeutics are medical devices considered a subset of digital medicine that deliver an independent therapeutic intervention and require clinical evidence or real-world outcomes. Regulatory oversight, not applied today, is needed.
Another barrier for digital medicine is the heterogeneous landscape of reimbursement within the various countries, with some more advanced than others. There is a complex landscape of disease management purposes that requires specific regulation approaches, such as well-being apps; remote monitoring; digital offerings for therapeutics and rehabilitation; and digital support for diagnosis, self-management education, and drugs.
Recchia closed with a future vision for the development of national digital health laws, stating that they will need to address the following topics: (a) doctors are allowed to prescribe medical apps; (b) improved access to patient data for research; (c) health care providers move patient communication and prescriptions to electronic channels; (d) every insured member has access to an electronic health record; (e) health insurers are allowed to offer online member signups; (f) health innovation is financially supported; (g) telehealth consultations become the norm; and (h) pharmacies, health care providers, and hospitals must connect to a secure communications network.
Cybersecurity
Enzo Tieghi, President, ServiTecno Srl, and Chair of the ISPE GAMP® Forum Italy, highlighted the need for cybersecurity. His main message was that cyber security is not only a matter of information technology, but also of operation technology because the digital transformation also changes the cyber-security perspective in operations. Within information technology, cybersecurity is necessary to protect data and information. Within operation technology, cybersecurity is necessary to protect systems that manage the controlled process.
Surveys indicate that after the energy sector, the health sector is the second-most targeted for criminal actions. Manufacturing is also in the top five targets. Thiegi outlined key pharma security breaches to know and learn from.
- 2014—Dragonfly malware attack: This cyberattack on the pharma supply chain was one of the first high-profile cyberattacks on the pharma industry.
- 2017—NotPetya malware attack on Merck: This attack is considered one of the most expensive and devastating in history. It affected over 30,000 com-puters, caused over $870 million in damages, and resulted in $410 million in lost sales.
- 2018–2019—Winnti malware attacks on Bayer and Roche: The purpose of these cyberattacks, thought to be linked to a state-backed Chinese hacking group, seemed to be industrial espionage.
- 2020—Dr. Reddy’s Laboratories data breach: The Indian drugmaker was forced to shut production facilities in the US, UK, Brazil, India, and Russia to isolate the data servers and address the cyberattack.
- 2020—EMA COVID-19 cyberattack: Pfizer, BioNTech, and AstraZeneca had vaccine-related information hacked when an EMA server was hit by a cyberattack.
- 2020–2021—North Korean cyberattack attempts: According to The Wall Street Journal, North Korean actors tried to steal vaccine information from Johnson & Johnson and Novovax, as well as three South Korean drugmakers.
Establishing a cybersecurity strategy can follow a maturity model with 129 questions (available at Servitecno) as presented. Subsequently he showed the Supervisory Control and Data Acquisition (SCADA) Security Maturity Model: Phase 1: Conduct assessments and define standards; Phase 2: Conduct training and raise awareness/funding; Phase 3: Deploy cybersecurity solutions to mitigate risks; and Phase 4: Operate and maintain. The proposed standards to be used are ISO 27001, ISO 27002, ISA 99, and IEC62443. Other success factors for implementation include budget owners, management, and leaders having a mechanistic understanding and the inclusion of industrial control systems (ICS) experts whose expertise can bridge the gap between ICS and IT security.
Cell and Gene Therapy
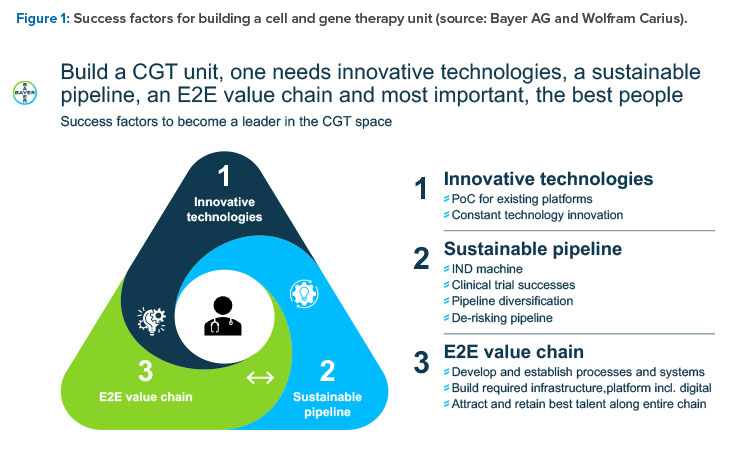
Wolfram Carius, Executive Vice President, Pharmaceuticals, Bayer AG, Germany, presented “How to Make the Biorevolution a Reality?” and discussed building Bayer‘s cell and gene therapy (C>) platform. What makes C> so attractive is the new disease intervention mechanism, which is distinct from pharmacotherapy, he said.
In the classical molecule world, intervention is at subcellular and cellular level by interaction with a molecular target, such as a receptor or enzyme. This treatment often leads to only symptomatic and needed permanent treatment. In the C> world, there is a direct interaction at the genetic level, with the opportunity to cure a genetic defect. Furthermore, modified cells are employed as curative agents (such as cytotoxic T-lymphocytes to kill cancer cells) or immunomodulatory cells to treat severe diseases (such as mesenchymal stromal cells against graft-versus-host disease). Degenerative diseases can be treated with engineered stem cells with specific features (such as secretion of cytokines to restitute or regenerate original function). The success is more often curative or regenerative rather than symptomatic or merely stopping disease progression.
In summary, C> is a multiproduct platform that allows for the restoration of biological functionality and can treat intractable diseases. It’s a multi-product platform that allows for several “shots on goal.” It offers restoration of tissue functionality in comparison to delay of disease progression (small molecules [SMOLs], big
molecules [BMOLs]) and enables treatment of previously intractable diseases. It’s a precision medicine with long-lasting effect (usually only one treatment is required) that allows high customization. It enables new curative potential by further enabling other processes; for example, through local pay-load (cytokines) delivery.
Such new products may need new concepts in quality assurance in logistics and in business models for payment, such as in the case of personalized medicines with short shelf lives, very high development cost, and batch sizes of only one unit.
Three organizational models for a C> unit were considered, from full integration in existing organization including all services and support to a complete standalone unit. At the end, a mixture of both was considered best to combine the advantages and minimize the downsides of the other “puristic” models.
Quantum Computing
Clemens Utschig-Utschig, CTO and Head of IT Technology Strategy, Boehringer Ingelheim, Austria, highlighted quantum computing and pharma. Quantum computing is a type of computation that harnesses the collective properties of quantum states such as superposition, interference, and entanglement to perform calculations. Utschig-Utschig named some use cases across the research and development value chain for quantum computing such as imaging of tissues, molecular dynamics situation, and binding affinity prediction.
In preclinical development, quantum computing can be used for side-effect prediction. In clinical development, it can be used for prediction of pharmacokinetics. However, to have a real quantum advantage in the pharmaceutical industry, there is a need to improve existing quantum algorithms, develop new quantum algorithms for many applications, and see the predicted hardware development to become reality and test heuristics. Basic research is required, but a hype could destroy quantum computing.
Gene Therapy
Frederic Revah, CEO, Genethon, France, focused on gene therapy. There are some main principles, such as in vivo gene therapy via direct administration, where recombinant adeno-associated virus vectors are directly injected into the target organ—for example, in the treatment of muscular dystrophies, eye disorders, liver disorders, hemophilia, or Huntington’s disease. Another principle is ex vivo hematopoietic stem cell transduction, where hemato-poietic cells taken from the patient are transduced with an HIV-derived lentivector and reinfused for the treatment of immune deficiencies, blood disorders, and CNS lysosomal storage disease. A third example is the cancer gene therapy based on stimulation of the immune response using chimeric antigen receptor (CAR) T-cells. CAR T-cells solve a basic problem of cancer therapy, the fight against tumors, which are invisible to the patient’s immune system. Gene-modified T-cells combined with synthetic antigene-specific receptors attack: Step 1 is sampling of patient T-cells; step 2 is ex vivo gene transfer into T-cells, allowing specific targeting to tumor cells and amplification; and step 3 is infusion of modified T-cells.
The challenge of manufacturing was summarized as “addressing the dose issue.” Revah showed the bioprocess innovation in six steps: (a) yield improvement × 100; (b) improved and novel producing cells, with super-producer cells adapted to a serum-free suspension method; (c) novel transfecting agents to improve transfection, decrease plasmid needs, and improve scalability;
(d) innovative plasmids that are a major cost factor by decreasing plasmid quantities; (e) innovative production platforms, including novel cell types and cell-free systems; and (f) analytical methods to improve accuracy of analyses for product and contaminants, facilitate online analysis, and decrease quantities required for quality control.
New EU Legislation
Nathalie Moll, Director General, European Federation of Pharmaceutical Industries Associations (EFPIA), listed the EU pharma strategy’s objectives, such as unmet needs and access, competitive and innovative industry, and resilience. Unmet needs and access means prioritizing unmet medical needs/antimicrobial resistance, ensuring patients’ access to medicines, and ensuring affordability of medicines for patients and health systems’ financial and fiscal sustainability. Competitive and innovative industry means providing a fertile environment for Europe’s industry, enabling innovation and digital transformation, and a sound and flexible regulatory system. Resilience means securing the supply of medicines across the EU and avoiding shortages; providing high-quality, safe, and environmentally sustainable medicines; and enhancing Europe’s health crisis response mechanisms.
The strategy adopted in November 2020 includes as one of its pillars: enhancing resilience, diversified and secure supply chains; environmentally sustainable pharmaceuticals; and crisis preparedness and response mechanisms. Key proposals are to revise the pharmaceutical legislation to enhance the security of supply, address shortages, and report on root causes of shortages in a structured dialogue. A European Health Union package targeted for the end of 2022 will contain a regulation that extends the EMA and European Centre for Disease Prevention and Control (ECDC) mandate, regulation on serious cross-border health threats, and a new Health Emergency Preparedness and Response Authority (HERA).
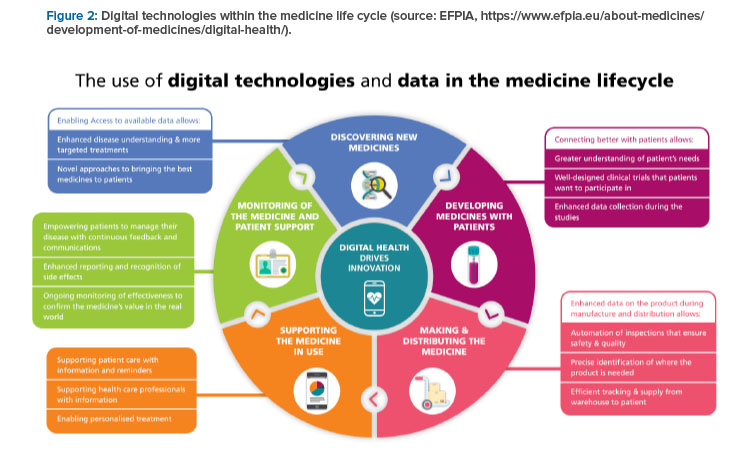
The EFPIA proposals to efficiently strengthen the supply chain include a common framework, a risk-based approach, and IT and flexibility. The common framework requires a common definition of a shortages and reporting standard and a harmonized reporting system with interoperable data. A risk-based approach requires focus on the most critical products as a combination of severity for the patient, the likelihood to be in storage and the availability of alternatives; robust shortage prevention plans; and stockpiling at the EU level. IT and flexibility require electronic platform leaflets and the European Medicines Verification System for shortage prevention and monitoring. All this will be supported by digital technologies, as indicated in Figure 2.
Innovative Manufacturing
Evdokia Korakianiti, Head of Quality and Safety of Medicines, EMA, Netherlands, led the session “Looking into the Future: Facilitating the Use of Innovative Manufacturing Approaches from an EU Perspective.” The EU vision on innovation has the following key drivers: accessibility and availability of medicines; data analytics, digital tools, and digital transformation; innovation; antimicrobial resistance; supply chain challenges; and sustainability of the network and operational excellence bodies.
The defined goals and objectives for innovation are to catalyze the integration of science and technology in medicines development in three ways. First, by supporting the integration of scientific and technological progress in the development of medicines (e.g., precision medicine, biomarkers, omics, and ATMPs) and ultimately into patient treatment. Second, by implementing an EU model for efficient, timely, and coordinated horizon scanning and priority setting that fulfills the needs of regulators, health technology assessment bodies, and payers. Third, by facilitating the implementation of novel manufacturing technologies.
Korakianiti highlighted the following as innovation trends for 2025–2030: (a) gene therapy and genome editing (in vivo gene editing); (b) microbiome products; (c) digital health; (d) vaccines using novel technologies (mRNA, viral vectors, and nano-delivery systems); (e) nanomaterials for targeted and modified release formulations; (f) novel manufacturing approaches (small portable manufacturing sites); (g) decentralized manufacturing (cell processing ATMPs); (h) 3D printing; (i) end-to-end continuous manufacturing (CM); (j) automation; (k) artificial intelligence and big-data approaches (Pharma 4.0™), and (l) individualized therapies.
The defined goals for supply chain challenges are fivefold. First, enhance traceability, oversight, and security in the medicine supply chain from manufacturing to importation and final use of active pharmaceutical ingredients (APIs) and excipients. Second, enhance inspector capacity, building at EU and international level to address the problem of APIs, new technologies, and continuous manufacturing. Third, reinforce the responsibility for product quality by harmonizing and reinforcing guidance to facilitate a coherent approach to the standards by regulators and industries for medicinal products. Fourth, encourage supply chain resilience and review long-term risks resulting from dependency on limited number of manufacturers and sites to ensure continuity of supply and availability of medicinal products. Fifth, analyze the possible implications of new manufacturing technologies and adapt the regulatory framework to accommodate innovation in manufacturing and distribution of medicinal products.
Pharmaceutical legislation needs to develop as well, Korakianiti explained, stating “there are no barriers in the legislation preventing advanced manufacturing but clarifications and guidance are needed.”
Korakianiti identified six main areas, with their hot topics and the regulatory environment:
- Decentralized manufacturing: Considerations for more flexibility for manufacturing and importation authorization licenses for remote sites, better define responsibilities and duties of QP and marketing authorization holders, and supervise central and remote sites.
- Personalized medicine: Clarify the adaptations and platform approaches permitted for such products (for example, constant part/adapted part). Consider aspects of traceability, data privacy, pharmacovigilance, and product information. Consider manufacturing steps that may be adapted and require data requirements to justify and support proposed adaptations. Verify adaptations in GMP principles to personalized medicines. Complete batch definition and batch release.
- Pharma 4.0™: Align expectations and definitions with other parts of the legal framework that deal with digitalization (e.g., artificial intelligence) such that it does not create a divergence. Update GMP chapters and annexes.
- Continuous manufacturing: Currently, there are five marketing authorization applications and one variation already approved. Enable adaptations or clarifications for batch concepts. Support end-to-end CM: GMP, API, and finished product specifications. Develop control strategy digitalization and modeling.
- General: Consider the life-cycle approach. Revisit the variation classification guideline (e.g., allow real-time changes in adaptative manufacturing processes and/or control strategies). Update assessor and inspector practices.
- Regulatory environment: Consider multiple voices. Watch for a lack of a single engagement pathway that follows through from proof of concept at the technology level through to product development, IMP, approval, and major related life-cycle changes. Consider the overall potential of a manufacturing innovation to influence many products or if the global supply chain is not easily built into the value proposition for a single product; this includes innovative methods increasing uncertainty, risk, and cost. Regional disconnect does not facilitate global development and manufacture. Consider regulator expertise capacity and culture toward risk.
The EMA is committed to creating a regulatory environment that fosters advanced manufacturing applications. The newly founded Quality Innovation Group of EMA is a key enabler as well as the international collaboration and efforts being made within the ICH and at the International Coalition of Medicines Regulatory Authorities (ICMRA) level.
Quality Surveillance
Nandini Rakala, Data Scientist and Visiting Associate, Office of Quality Surveillance, Office of Pharmaceutical Quality, CDER, FDA, spoke about advancing quality surveillance. Five key areas have been highlighted: pharmaceutical quality system effectiveness, quality signal detection and topic modeling post-market quality surveillance, risk-based prioritization using machine learning, quality metrics, and quality management maturity.
Pharmaceutical Quality System Effectiveness
The strategy is to integrate advanced analytics into quality surveillance to enhance an overall proactive quality surveillance framework through the integration of predictive analytics and artificial intelligence (AI)-based machine learning (ML) techniques. Subjects will be quality metrics, quality management maturity, sites engagement program, and ongoing research exploring applications of ML to improve site selection models.
The pharmaceutical quality system (PQS) assessment framework is related to the facility (inspection and product quality defect reports) and to the historical data for performance metrics (corrective and preventive action [CAPA] effectiveness, investigation times, human error, root cause, repeat deviation, and time to initiate recall).
Assessed qualitative elements are management commitment, quality policy, quality planning, resource management, internal communication, management review, management of outsourced activities and purchased materials, process performance and product quality monitoring system, CAPA systems, change management, and continuous improvement. Modeling a pipeline for data flow and the creation of new consolidated performance metrics can lead to predictive scoring and benchmarking illustration. All data can be used!
Quality Signal Detection and Topic Modeling Postmarketing Quality Surveillance
The Office of Quality Surveillance requires a systematic and data-driven approach to identify potential quality signals in postmarket surveillance reports. The objectives are to first, identify changes in the reporting habits and product names to prioritize resources and identify potential product quality signals; and second, create a function that uses statistical process control charts to flag entities with unexpected changes in their quality defect reporting habits. In addition, there is the application of natural language processing (NLP) algorithms to discover topic clusters and commonly occurring problems across the network of regulated facilities and products.
Risk-Based Predictive Prioritization
Field alert report (FAR) prioritization is an innovative AI framework leveraging ML and NLP for risk-based prioritization of incoming initial FARs. It is applied using multidisciplinary analytics and subject matter expert collaboration by incorporating input on data cleaning, topic-keywords refinement, ra-re-events tagging, and risk-based target creation in a programmatic manner. There is a streamlined process of data preprocessing and developing topic model predictors, merged with other key indicator variables for predictive scoring. Insights generated by this process will be used to proactively prioritize assignments and inform key indicator variables interpreted and recommended by the data-driven machine learning hybrid model.
Quality Metrics
The quality metrics program objectives are to analyze the quality metrics data to obtain a more quantitative and objective measure of manufacturing to ensure quality and reliability. The idea is to integrate the metrics data and resulting analysis into the FDA’s comprehensive quality surveillance program. Then, apply the analysis results to assist in identifying products at risk for quality problems and to enable sustainable current good manufacturing practices (CGMP) compliance supported by continual improvement, promote an effective PQS, and mitigate drug shortages. Furthermore, the strategy is to develop compliance and inspection policies and practices to improve the agency’s ability to predict future drug shortages. It is intended to be a mandatory program.
Key lessons learned from a feedback program included that cross-sectional analysis focused on comparing sites and products is not meaningful without context. It’s more appropriate to evaluate site performance over time for trends, shifts, and change points. In some instances, a combination of metrics rather than a single metric was preferred to assess a particular practice area.
The FDA’s analysis of the data submitted indicated the applications of statistical quality control, ML, and NLP as appropriate, and advanced analytical techniques used to assess quality metrics data submitted by industry. FDA learned that pilot firms are also deploying these techniques to monitor performance and identify signals. Certain metric calculations based on the definitions from the 2016 revised draft Quality Metrics Data guidance can result in mathematical discrepancies that are caused by inherent variabilities from real-time operations. PQS effectiveness is a critical component of a metrics program, as evidenced by numerous sites submitting data around their CAPA program, repeat deviations, and other timeliness metrics.
The proposed approach for a quality metrics reporting program includes manufacturing process performance, which includes process capability/performance indices, right-first-time rate, and lot release cycle time; PQS effectiveness, which includes CAPA effectiveness, repeat deviation rate, change control and equipment effectiveness, and unplanned maintenance; laboratory performance, which includes adherence to lead time, right-first-time rate, and calibration timeliness; and supply chain robustness, which includes on-time in-full, fill rate, days of inventory on-hand, and dis-position on time.
Quality Management Maturity (QMM)
QMM is defined as a state attained by having consistent, reliable, and robust business processes to achieve quality objectives and promote continual improvement. Meeting CGMPs is the minimum standard for legally marketing drug products in the US. CGMPs assure proper design, monitoring, and controls for manufacturing processes and facilities. Fully realizing the pharmaceutical quality vision for the 21st century requires moving toward richer quality management systems.
QMM is not a discrete concept: it is a holistic concept that includes PQS effectiveness and many related aspects of business processes that enable manufacturers to proactively monitor quality-related events. Driven by management commitment, manufacturing strategies, supply chain management, quality risk management, and effective knowledge management, it results in targeted, prioritized, and risk-based mitigation plans driven by data and advanced analytics. QMM is intended to be a voluntary program aimed at recognizing and rewarding manufacturers for “mature quality systems” that achieve sustainable compliance and focus on continuous improvement, business continuity plans, and early detection of supply chain issues.
All stakeholders with oversight and controls over manufacturing take ownership for quality. Management sets the tone of commitment to quality and drives the budget, adoption, and integration of quality. Organizational objectives drive quality, thereby reducing cost of quality. Quality systems shape the manufacturing site’s culture. These stakeholders should prioritize investing in people, designing an optimized patient-centric experience from the outside in, moving toward a performance-based QMM, and focusing on innovation and continual improvement, with a strong sense of change and knowledge management. They should support proactive risk management, mitigation, planning, and forecasting using application of predictive analytics and state-of-the-art optimization. Further, they should ensure there is a robust metrics program in place, driven by advanced analytical methods, sophisticated statistical tools and techniques, and AI-based technologies augmented by human intelligence. ICH Q10 and cGMPs are basic requirements for a PQS. Quality metrics are a key aspect of a mature PQS, using data-driven approaches to reduce quality issues and drive continual improvement.
Building a QMM program is bolstered by the data learned from efforts to date, such as the PDA Quality Culture Initiative, ISPE Advancing Pharmaceutical Quality Program, Quality Excellence University of St.Gallen, FDA/CDRH Case for Quality Pilot Program, Dun & Bradstreet Quality Benchmarking Study and others. The FDA initiated a QMM pilot assessment framework with six program areas: leadership and governance, operations, continual improvement, stakeholder engagement and satisfaction, knowledge management, and workforce engagement. The framework also has four pillars: sustainability, risk management, compliance, and quality culture. The outcome of these pilots have been published in an FDA white paper titled “Quality Management Maturity: Essential for Stable U.S. Supply Chains of Quality Pharmaceuticals”.1
The insights generated will be used to enhance comprehensive surveillance through proactive and risk-based data-driven decision-making. Current research indicates quality metrics as a key aspect of a mature PQS. Both strong metrics and quality culture programs are part of QMM, which assists in reducing the risk of supply chain disruptions and in fostering a culture of high quality. The FDA envisions that a QMM rating system will provide a more robust drug supply chain and greater commitment to quality in the pharmaceutical manufacturing industry, thereby leading to reduced potential for drug shortages.
Conclusion
The keynotes and sessions provided a wealth of information to attendees. Over 90% of the feedback collected after the conference rated the event as very good or excellent. This was primarily related to the quality of presentations, speeches, and panel discussions. The first in-person conference in years brought members and speakers together for an impactful event.
About the Author



