Quality Risk Management: Ensuring Shared Responsibility across Key Stakeholders

ISPE’s second virtual Global Pharmaceutical Regulatory Summit, held on 16 June 2021, brought together regulators from European Medicines Agency (EMA), Australian Therapeutic Goods Administration (TGA), US Food and Drug Administration, and Center for Drug Regulation and Research, FDA Philippines to share their insights on application of quality risk management. They discussed the positive impact and best practices for collaboration and shared responsibility between regulatory agencies and industry, which depends not only on consistent processes and protocols, but more importantly on the commitment of all key stakeholders. An industry perspective was included in the panel discussion from two senior leaders, one each from Abbvie and Cipla.

Participants were






Brendan Cuddy led off the program started by reminding the audience of the importance of International Conference on Harmonization (ICH) guideline, Q9, Quality Risk Management, which was signed off in November 2005. In combination with other ICH Guidelines Q8, Pharmaceutical Development, Q10, Pharmaceutical Quality System and Q11, Development and Manufacture of Drug Substances, Q9 had resulted in major efforts to modernize the GMPs and reflect risk-based thinking in many areas of the pharmaceutical industry. There have been many good initiatives undertaken globally since 2005. Some European examples of new guidances, which include better understanding and application of risk management are:
- EU GMP Guide, Annex 1, Manufacture of Sterile Medicinal Products, which is currently undergoing a major update
- Reflection Paper on Good Manufacturing Practice and Marketing Authorisation Holders
- EU GMP Guide, Annex 4 and Annex 5 on Manufacture of Veterinary Medicinal Products
In practice, regulators have found during GMP inspections and in regulatory applications that QRM principles are applied inconsistently. Both industry and regulators would benefit from a limited revision of ICH Q9. This revision would improve the understanding and consistent application of QRM by addressing subjectivity in risk assessment/QRM outputs, supply and product availability risks, formality in QRM and risk-based decision making.
Regulators are pulling together information from inspection, data from market surveillance and quality defect root cause analysis to build knowledge to impact improved risk-based Inspection Planning, Market Surveillance testing and classification of quality defects and root cause analysis. Additionally, the PIC/S Expert Circle on QRM is developing inspector training.
Cuddy continued by discussing the exceptional approaches needed during the recent pandemic to ensure that ongoing verification of GMP compliance could be maintained. Authorities must retain the responsibility to assess and verify compliance. Distant assessments can represent a suitable means of determining compliance with the principles and guidelines of GMP/GDP. Guidance related to GMP/GDP distant assessments is available on the EMA web site.
Examples of regulatory flexibilities applied by European Commission/EMA/Heads of Medicines Agencies (HMA) until the end of the Covid pandemic are:
- Questions and Answers on Regulatory Expectations for Medicinal Products for Human Use During The Covid-19 Pandemic (https://ec.europa.eu/health/sites/default/files/human-use/docs/guidance_regulatory_covid19_en.pdf) developed by the EMA Good Manufacturing Practice and Good Distribution Practice (GMDP) Implementation Working Group (IWG)
- Automatic extension of validity date of GMP certificates that can be used In regulatory submissions
- Product specific GMP flexibilities for crucial medicines
- Non-product specific GMP and GDP flexibilities
Cuddy discussed challenges amplified during the pandemic such as:
- The need for treatments and building manufacturing capacity for Covid medicines
- Medical innovations
- Global nature of development and manufacturing
- Need for globally cooperation between regulators, e.g. on post approval changes
He asked if lessons learned from the pandemic could lead to more agile and resilient supply chains within an agile and flexible regulatory system. He also asked whether there is a future for leaner post-approval change management in terms of:
- Harmonized data standards and descriptive information,
- Convergence and harmonization in assessment and inspections,
- Reliance in assessment and inspection, and
- Realizing the benefits of ICH Q10 and ICH Q12, Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management
Cuddy recognized that extensive work will be required in international fora with all stakeholders to bring all these about.
Cuddy reminded the audience that managing risk everyday depended on people and culture, however, what matters is that risks and threats are methodically and objectively interpreted rather than being downplayed or concealed.
Ben Noyen presented on TGA experiences of remote GMP inspections and how sites should prepare for them. From a TGA perspective, the pandemic has impacted:
- The ability to perform routine on-site inspections
- Increased inspection complexity
- Potential for reduction of routine GMP signals
- Required increased collaboration with mutual recognition agreement (MRA) partners, particularly regarding acceptance of distant assessment certificates.
TGA has a well-developed approach to GMP inspections, which prior to the pandemic included a desk top evaluation. As a result of the pandemic, virtual or hybrid inspections have been introduced, with potential cooperation with a local regulatory agency if the manufacturer is overseas as part of the overall process. TGA would like remote inspections to continue and would welcome more harmonized tools to facilitate reliance between agencies.
These new arrangements are needed to:
- Minimize potential impacts from an on-site inspection for both industry and TGA staff
- Help continue ongoing governance of GMP inspections at domestic facilities
- Facilitate new GMP licenses and/or variations of licenses for existing products
- Maintain patient and consumer confidence
The process must use a risk-based model to evaluate the various options available to inspectors on a case-by-case basis in consultation with each manufacturing site. To determine the criticality of the type of inspection, the following main factors are considered:
- Criticality of product to market, manufacturing risk and manufacturer’s compliance history
- Levels of protection from Covid required of products and personnel, both manufacturing and inspection
- Manufacturer’s technical capability to support a remote inspection
To be ready for an inspection the manufacturer must have a completed site for which they have functional control and have implemented and documented a pharmaceutical quality management system.
The manufacturer’s staff play an important role in a remote inspection. They must have appropriate knowledge of the product(s) and manufacturing and quality operations. Clearly staff must be informed regarding the type and extent of an inspection, and where applicable, translators must be available. There is a need to build trust between inspectors and those being inspected.
Important technology considerations for manufacturers are:
- Availability of internet and bandwidth
- Functionality to share documents, images and pre-recordings and upload them
- Live streaming capability
During remote inspections manufacturer’s staff must be aware of the technology and its use and should have the ability to do some troubleshooting.
Noyen highlighted common compliance issues with the application of quality risk management (QRM) by manufacturers:
- Poor definition of when to apply QRM
- Poor training with respect to QRM processes
- Incomplete identification, assessment, and control of risk
- Inappropriate application of risk mitigation strategies
- Inappropriate acceptance of risk
- Poor choice of QRM tools
- Lack of awareness of risk processes and their outcomes
- Poor assessment of the effectiveness of risk mitigation strategies applied
- Compilated risk assessment processes that appear to be incorrectly applied and often not actioned
Other common inspection deficiencies are:
- Poor cross-contamination controls
- Poor management of quality incidents
- Insufficient evidence to support practical training of personnel
- Inadequate facility and equipment qualification, and process validation
Data integrity can be a challenge at remote inspections, particularly the control of computerized systems with respect to:
- No up-to-date listing of computerized systems and their GMP functionality
- Inadequate user access control
- Poor control of data on standalone systems
- Issues with audit trail assessments
- Control of third-party suppliers of cloud services
- Computer systems not validated for intended use.
Noyen made reference to the excellent series of guides available from the ISPE GAMP™ series.
Noyen concluded by asking how it may be possible to reward manufacturers with a good quality management system and better culture?
Joyce Cirunay, Director reminded the audience of Figure 1 in the ICH Q9 Quality Risk Management guideline, overview of a typical quality risk management process.
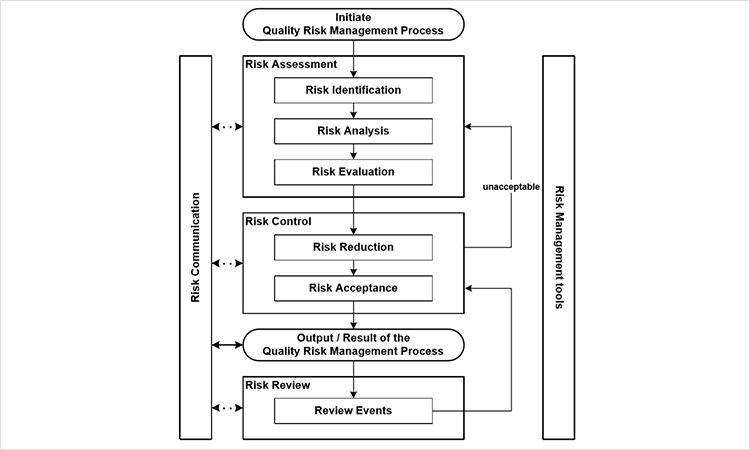
Cirunay continued with the definition of QRM as:
A systematic process for the assessment, control, communication and review of risks to the quality of the drug product across the product lifecycle.
Key Principles are:
- Evaluation of the risk to quality should be based on scientific knowledge and ultimately link to the protection of the patient
- Level of effort, formality and documentation of the quality risk management process should be commensurate with the level of risk.
Potential applications of QRM are in:
- Integrated quality management
- Regulatory operations
- Development
- Facilities, equipment and utilities
- Material management
- Production
- Laboratory control and stability studies
- Packaging and labelling
The legal basis for QRM is embedded in Philippines’ legislation. Examples of implementation exist in GMP inspections both pre-approval and post-approval, for example for:
- Risk-based GMP inspection planning
- Site selection process for GMP inspection
Pre-approval risk management is being applied for issuance of licenses to operate and for new drug applications. In dossier evaluation, low risk activities are post-approval changes and renewal applications whereas high-risk activities are vaccine, biologics’ and new drug dossier evaluations. There are further opportunities post-approval with product recall, routine reporting, labelling and pharmacovigilance.
Other opportunities for applying risk management in the Philippine regulatory processes are with elimination of unnecessary requirements.
David Churchward, Deputy Unit Manager, Inspectorate Strategy & Innovation (Expert GMP Inspector), MHRA then chaired a discussion with a panel consisting of the regulators and industry representative.
Amongst a series questions, the Panel addressed the following:
- What are the current challenges in supply to global markets – are these different to pre-pandemic?
- What are the current challenges of regulating a global supply chain – are these different to pre-pandemic?
- In the spirit of ‘shared responsibility’ to patients, how can we overcome these challenges?
- As representatives of international organisations with global manufacturing supply chains, is it possible to implement a truly global approach to a quality system? What insight can you provide that ensures successful delivery?
- What are some of the biggest challenges to implementing and communicating a QRM approach?
- GMP inspectors frequently cite QRM as a contributory factor to inspection observations. Is there a disconnect between industry implementation and regulatory expectations?
- What key indicators of a compliant QRM system are you looking for? What can regulators do to facilitate transparency?
- Shared responsibilities require transparency. How can this be achieved? Do industry and regulators need to change their behaviour?
- The pandemic has provided important lessons in collaboration (e.g. rolling review, closer engagement). What should be taken forward, and how?
The lively and interesting Panel discussion was concluded with closing remarks from Tom Hartman, President and CEO, ISPE. Hartman summarized the successful meeting, noting:
- Not all the challenges observed are “bad”. There are positive learnings for the future
- Embracing regulatory flexibilities has been successful
- Risk should be applied to decisions on supply of medicines
- Transparency and good communication are important to build trust and a better culture
- Reliance between regulators has been important
Disclaimer
This is brief and informal synopsis of a discussion among regulators from various countries and regulatory organizations during the ISPE Global Pharmaceutical Regulatory Summit on 16 June 2021. It has not been vetted by any of the agencies or regulators cited in this article.
About the Author



