EMA Support for Innovation in Pharmaceutical Manufacturing

At the 2025 ISPE Europe Annual Conference held 12-14 May 2025 in London, the European Medicines Agency (EMA)’s Head of Quality and Safety of Medicines Department, Evdokia Korakianiti, highlighted the many areas of support that the European Union (EU) is giving to promote innovation in pharmaceutical manufacturing. She outlined the regulatory framework and priorities, the EMA’s support mechanisms, key current initiatives and activities to promote international harmonization. The drivers and challenges to reform in the EU are:
- Uncertainty due to economic pressures on healthcare systems
- Affordability
- Availability by preventing drug shortages and securing supply
- Access for patients across the EU
Regulatory Framework and Priorities
There is ongoing revision of EU pharmaceutical legislation to provide better connected regulatory landscape. This is being finalized for adoption at the end of 2025 and implementation at the end of 2028 or early 2029. There are elements in this legislation which should catalyze innovation such as the definition of personalized medicines, provisions for decentralized manufacturing as well as use platforms.
Complementing this legislation reform is the Critical Medicines Act issued in March 2025 to ensure that all EU citizens have access to necessary medicines. This act is an output from the EU pharmaceutical strategy for Europe and seeks to provide support to the pharmaceutical industry in promoting research, innovation, and technologies that meet the therapeutic needs of patients while ensuring affordable access to medicines for patients. Additionally, the EU has introduced actions to boost biotechnology and biomanufacturing in the EU, which are a response to the Draghi report on EU competitiveness and which discuss a new competitiveness model based on innovation-led productivity. All these initiatives encourage regulators, for example, to support manufacturers to progress innovative technology.
EU regulators recognize the value of advanced manufacturing to improve assurance of quality, give agility and flexibility with the aim of efficiency and cost reduction. Regulators will engage with industry and advise medicines developers to enable the introduction of innovation. For example, regulators are contributing to the EU Flexible Assembly of Bio-manufacturing (FAB) ambition to have sufficient and agile manufacturing capacities for different vaccine types (mRNA-based, vector-based, and protein-based). These capacities will be kept operational and can be activated quickly in case of a public health emergency.
Korakianiti summarized EU and EMA support tools for innovation, for example:
- Innovation task force as a platform to facilitate innovation
- An office to support small and medium sized enterprises (SMEs)
- Quality Innovation Group (QIG), which is an operational expert group set up to support the translation of innovative approaches to the design, manufacture and quality control of medicines, to bring new therapies, and help improve the supply of existing medicines to patients
Regulatory pathways to progress accelerated development and approval in the EU include the Priority Medicines (PRIME) scheme, from which 28 medicines have been have been given accelerated assessment and authorized. There are 30 additional medicines in the pipeline. In addition, conditional marketing authorization provides early access to patients for medicines that address unmet medical needs. In these cases, conditional access may be granted early with reduced levels of clinical and non-clinical data based on favorable benefit compared to the risk.
The EU biotech and biomanufacturing hub supports companies, particularly SMEs and start-ups with regulatory and legislative guidance to achieve marketing authorization (MA) in the EU as well as potential financial opportunities.
Korakianiti stressed the success of the QIG. It consists of eight core members who represent regulatory expertise in chemical, biological, advanced therapeutic medicinal products (ATMPs), and good manufacturing practice (GMP) standards. In addition, the QIG has access to European experts, who are nominated by member states or EMA and made available by European national competent authorities or academic institutions from the member states of the European Economic Area (EU member states plus Iceland, Liechtenstein, and Norway), as part of the European Medicines Regulatory Network (EMRN). It is an open forum for discussion of innovations from SMEs, large companies, academics, and vendors where all stakeholders come together to discuss the opportunities and challenges. For many meetings the QIG invites regulatory partners to attend, such as the US Food and Drug Administration (US FDA), Japan’s Pharmaceuticals and Medical Devices Agency (PMDA) and the UK’s Medicines and Healthcare products Regulatory Agency (MHRA). The QIG provides advice throughout the product lifecycle on regulatory approaches to assessment and inspection, and as well as preparing regulatory guidance on innovative quality and manufacturing technologies. In addition, the QIG has hosted five listen and learn focus group meetings (LLFGs) on the topics of:
- Decentralized manufacturing
- Continuous manufacturing
- Digitalization and automation of manufacturing
- Process models
- Platform manufacturing technologies
- Personalized medicines
Case studies are presented and discussed with challenges and potential solutions proposed and concrete actions identified to give connected and predictable advice from proof of concept through the product lifecycle. Reports are issued and available on the QIG web site.
The QIG’s achievements are summarized as:
- Scientific advice to five EU programs, e.g., drug shortages, improving supply capacity, and reducing manufacturing costs
- Seventeen one-to-one meetings and three site visits, e.g., decentralized medicines, digital twins, 3-D printing
- Support to 33 innovation task force meetings, e.g., process automation, continuous manufacturing for biologics, artificial intelligence applications
- Support to guidance preparation including guidance being drafted by regulatory partners, e.g., considerations on process models, reflections on decentralized manufacturing, contribution to EU GMP Annex 11 on computerized systems, and 22 drafts related to artificial intelligence (AI)
Korakianiti expanded on some of these EU and EMA initiatives aimed to foster innovation and accelerate access to new medicines.
The EU is very active in promoting and guiding the introduction of more digitalization and use of AI in pharmaceutical manufacturing. The EU Artificial Intelligence Act, 2025, is the first regulation on AI and it is designed to make sure that AI systems used in the EU are safe, transparent, traceable, non-discriminatory, and environmentally friendly. AI systems should be overseen by people, rather than by automation, to prevent harmful outcomes. It also establishes a technology-neutral, uniform definition for AI that could be applied to future AI systems.
EMA issued a “Reflection paper on the use of AI in the lifecycle of medicines” in 2025 as part of the Big Data Workplan 2022-2025 to develop the EMRN’s capability in data-driven regulation. This is a risk-based approach for development, deployment and monitoring of AI and machine learning (ML) tools.
The QIG has started preliminary considerations on use of process models, for example by holding a listen and learn session.
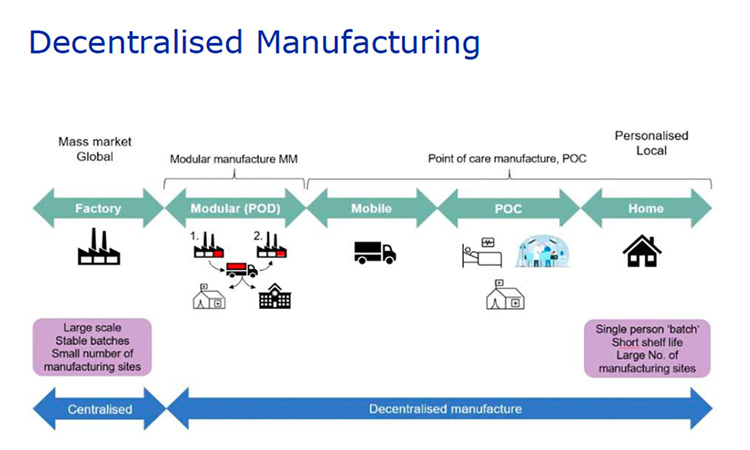
Text is being drafted for the EU directive related to implementation of decentralized manufacturing. This initial text is focused on products, e.g. ATMPs with a short shelf life which require manufacture and/or testing close to the patient. The aim is that multiple point of care (POC) manufacturing sites should not require a separate manufacturing authorization from the one granted to the relevant central site as shown in the screen shot from the EMA slide presented by Korakianiti given below.
- Korakianiti discussed the role of prior knowledge as a data baseline, which can be used to accelerate development, dossier preparation and review. It has been used for COVID-19 vaccines, monoclonals and oligonucleotides. It does require data to be placed in context, explained and justified.
- The concept of platform technology is similar to but also different from prior knowledge. What constitutes a platform, how it can be used in regulatory applications, updated and future-proofed is under consideration for inclusion in the upcoming new EU pharmaceutical legislation. The aim is a regulatory mechanism which can be cross referenced such that development can be optimized and dossiers can refer to it obviating redundant work and speeding development and review.
- In the field of personalized medicines new legislation is being drafted to recognize that a medicinal product may have a fixed component and a variable component tailored to make the product suitable for a patient or group of patients. Examples of the variable component could be starting materials, raw materials, manufacturing processes, administration patterns, or doses. Medicines being considered within scope are individualized immunotherapy against cancer and individualized bacteriophage cocktail against infection.
Korakianiti stressed the EU’s commitment to global collaboration to agree a common set of rules and approaches to find new ways of working to solve global health challenges. She outlined collaborations with the European Commission, the World Health Organization (WHO), the US FDA, and the International Coalition of Medicines Regulatory Authorities (ICMRA). Specific international efforts are to simplify lifecycle management, i.e. post approval applications. There has been an ICMRA pilot on assessment of post approval change management applications and on hybrid inspections. The Post Approval Change Management Protocol (PACMP) process involved multi-agency collaborative assessment of identical submissions in parallel with the aim to have a harmonized outcome. This pilot is being reported as a success and is being continued. Therefore, further participation is requested; hence companies interested should apply via the ICMRA website.
EMA is also involved in the WHO post-approval reliance pilots, which is applicable to Type II (pre-approval) applications. This is a two-stage process with an initial EMA assessment and outcome shared with participating agencies. The aim is to achieve shorter approval times across participating global regions.
Conclusion
Key learnings for EMA from introduction of recent regulatory initiatives are:
- Different levels of technology maturity require different levels of support
- Early dialogue with developers and other international regulatory partners is extremely beneficial
- Flexibility is required for example developing agile guidance documents
- Knowledge sharing with developers and international regulators is very important
- Continued regulatory convergence across the globe is required
Korakianiti concluded that EMA is a partner in innovation and supports developers from bench to bedside, and its doors are open. Innovation benefits patients worldwide.
Learn more about ISPE’s regulatory initiatives and programs.
Find out about upcoming ISPE conferences.
Disclaimer:
This is an informal summary of a presentation on 14 May 2025 at the 2025 ISPE Europe Annual Conference in London. It has not been vetted by any of the agencies or regulators mentioned in this article, nor should it be considered the official positions of any of the agencies mentioned.
About the Author



