Continuous Manufacturing in Biotech Processes - Challenges for Implementation

Continuous biotech manufacturing has the potential to achieve greater product quality at lower cost and shorter time to market. Recent advances in fully integrated control systems and a dynamic market make it more relevant than ever.
Continuous processes have become standard in many industries because they provide the best use of installed assets and produce products of consistent quality. They have been implemented in the production of pharmaceutical and food ingredients. Although continuous manufacturing (CM) in the biotech industry has been discussed for many years, recent advances in fully integrated control systems combined with the dynamic state of the market make it more relevant than ever before.
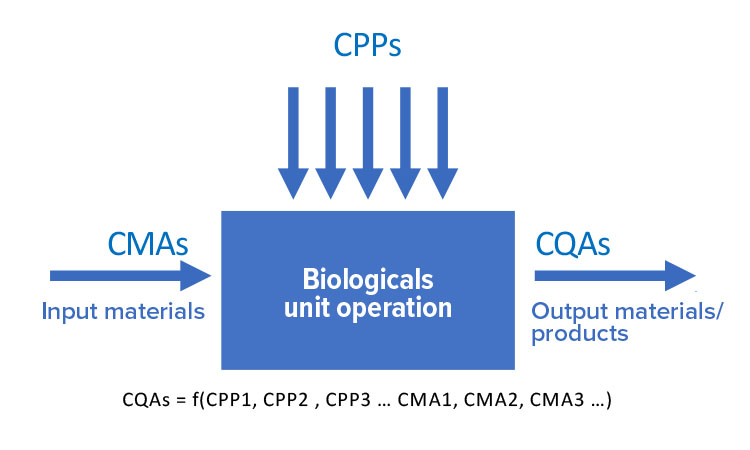
Continuous processes are operated by automated systems in which stable critical quality attributes (CQAs) are achieved by adjusting critical process parameters (CPPs) in real time. This improves process robustness, productivity, and equipment utilization. Yet even though the scientific knowledge to develop continuous processes is available, batch processes are still used almost exclusively to manufacture biopharmaceutical products. The biotechnology industry requires a shift in this mentality.
CM has many advantages: The time required for development is shorter, since multiple sets of parameters can be evaluated during a single lot. Less human intervention is required, reducing operational cost and human error. CM has a smaller footprint. By integrating single-use technology into the process line, initial capital investment may be reduced as well. Lastly, pilot-scale processes, which may also be used for clinical supply, can transition into commercial manufacturing simply by increasing the run time. This eliminates scale-up and its associated construction and validation steps and speeds up time to market. Since it is sometimes difficult to predict market demand when sizing the ¬first commercial batch to scale process, the flexibility of continuous processes mitigates the risk of making the wrong assumption.
With all these advantages, it is no surprise that the development of CM in biopharmaceuticals has generated growing interest in the scientific community and spurred increased investment by drug manufacturers. Continuous processes have been implemented in the production of pharmaceutical and food ingredients. Continuous purification technology is commonly used for commercial production. Continuous perfusion (cell culture) processes have also been used for several years. Technology for the remaining upstream and downstream unit operations is coming to market.
Because many challenges remain to be overcome before its adoption, this article will present the status of CM in the commercial product lifecycle and outline work that has yet to be completed:
- R&D efforts to characterize CPPs on single-unit operations and integrate them into the process
- Integrated control strategy required for operation
- Technology available on the market
- Approaches to validation and quality
- Regulatory compliance
R&D Challenges
This section lists continuous processing challenges in R&D and proposes a workow and tools for future development work.
A continuous mode needs single-unit operations, but must also master the interplay between them. As Konstantinov and Clooney note in their whitepaper (emphases in original):
- A unit operation is continuous if it is capable of processing a continuous ¬ ow input for prolonged periods of time. A continuous unit operation has minimal internal hold volume. The output can be continuous or discretized in small packets produced in a cyclic manner.
- A process is continuous if it is composed of integrated (physically connected) continuous unit operations with zero or minimal hold volume in between [ and suitable controls are in place to capture process variations].1
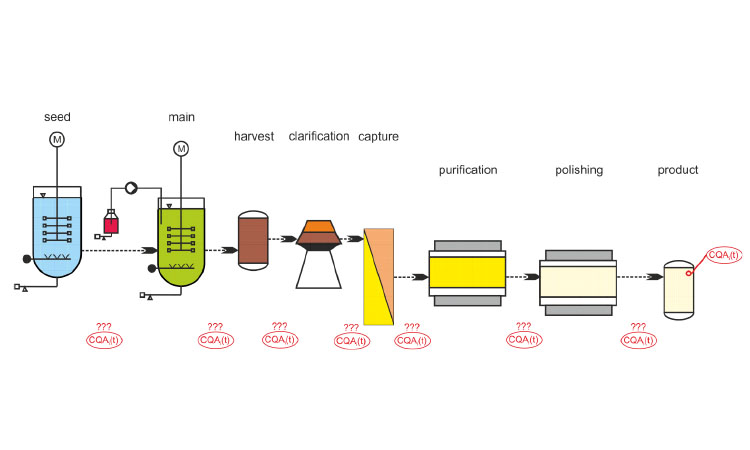
This differentiation of continuous unit operations and continuous integrated processing (Figure 1) dictates the development steps: 2
- Understand and establish single-unit operations
- Link them to an integrated process
Continuous processing requires a different level of process understanding. As an example, classical antibody production, which uses Chinese hamster ovary (CHO) cells as a host, pools the product solution after 14 days. The time-variant dependency of CPPs and CQAs are integrated in one analytical result and the process is also registered as such. Hence, in batch processes, the outcome of the current step determines the subsequent step. When moving to continuous operation, we have to understand the time dependency between CPPs and CQAs. This calls for the enhanced use of metabolic and kinetic models and different experimental designs.
For example, classical designs of experiments (DoEs) can only capture the response of the system to time-invariant factors. For continuous operation the development strategy is reversed. We want to run with time-invariant process variables, but we need to understand the time-variant dependencies on their CPPs for control. We may need dynamic model-based DoEs to develop a control strategy able to cope with process variability over time; those experimental setups should be established in the R&D environments.
| Unit operation | Design principle |
|---|---|
| Media and buffer preparation | Continuous |
| Cell culture | Continuous |
| Chromatography | Staggered |
| Filtration | Continuous |
| Viral inactivation | Staggered |
Continuous downstream processing starts in robust continuous upstream processing. Continuous downstream processing systems that can achieve steady-state operations may be soon available. Before we can achieve that, however, we will also need continuous upstream solutions. Since the cell lifecycle goes from exponential growth to producing, necrotic, apoptotic, dead, and lysed states, current perfusion processes, which monitor only viable cell count and viability, may not be enough to understand the dynamically changing population. Further analytical techniques, such as fluorescence-activated cell sorting and population models, must be applied.
A more promising approach uses metabolically balanced cells in which the primary and secondary metabolism prevails to the metabolic burden of the recombinant product. For this, tunable promoters with genetic integration and low metabolic burden are preferred over the constitutive promoters3 currently used in CHO cell line development.
Continuous processing requires a much earlier definition of the process control strategy. As proposed by current US Food and Drug Administration (FDA) validation guidelines, stage 1 validation includes process characterization studies (PCSs), which are the “collection and evaluation of data, from the process design stage throughout production, which establishes scientific evidence that a process is capable of consistently delivering quality product.”4
PCSs reveal the interplay of process parameters (PPs) on CQAs, demonstrate process robustness within multivariate normal operating ranges (NORs), and propose the control strategy, including process and analytical controls. This is currently achieved for batch processes by fusing development and manufacturing data.
For continuous processing, time-variant dependencies between CPPs as CQAs must be transformed in a control concept, which we need as prerequisite for process design. NORs must also be defined earlier as part of a process control strategy, based on process analytical technology (PAT) models and controls. This means that PCSs should be completed during development. These may include data mining, risk assessments, characterization of process performance, screening studies, criticality assessment,5 and integrated process modeling.6 Scale-down model qualification may not be necessary, as the development scale may be already the production scale, since productivity is scaled by processing time. R&D labs, on the other hand, need greater data management and data science orientation skills.
- 1Konstantinov K. B., and C. L. Cooney. “White Paper on Continuous Bioprocessing.” May 20–21, 2014 Continuous Manufacturing Symposium. Journal of Pharmaceutical Sciences 104, no. 3 (March 2015): 813–20.
- 2Rajamanickam, V., et al. “Impurity Monitoring as Novel PAT tool for Continuous Biopharmaceutical Processes.” Continuous Bioprocessing: A Repligen E-Book. Industry best practices compiled by Repligen. 2016. https://www.repligen.com/landing-pages/continuous-bioprocessing-book
- 3Wurm, D. J., et al. “Mechanistic Platform Knowledge of Concomitant Sugar Uptake in Escherichia coli BL21 (DE3) Strains.” Scientific Reports 7 (2017): 45072.
- 4US Food and Drug Administration. Guidance for Industry. “Process Validation: General Principles and Practices. January 2011. https://www.fda.gov/downloads/drugs/guidances/ucm070336.pdf
- 5Zahel, T., et al. “Workflow for Criticality Assessment Applied in Biopharmaceutical Process Validation Stage 1.” Bioengineering (Basel) 4, no. 4 (December 2017): 85. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5746752
- 6Zahel, T., et al. “Integrated Process Modeling—A Process Validation Life Cycle Companion.” Bioengineering (Basel) 4, no. 4 (December 2017): 86. ,a href="https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5746753">https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5746753
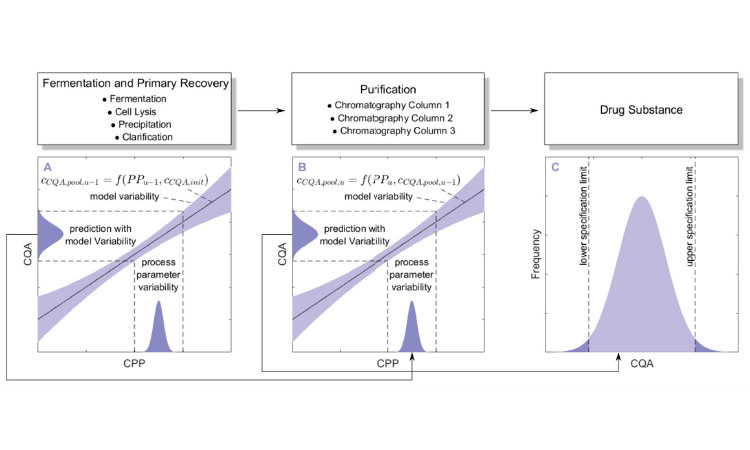
Using integrated modeling to accelerate continuous process development. In addition to understanding single-unit operations, the interplay between them is an important component of robust processing. Integrated process models (e.g., ASPEN, G-Proms) have been used successfully in other market segments and could be applied to batch or continuous bioprocesses to identify PPs and critical control strategies. These models should display the process and NORs of individual unit operations,5 ,6 link individual unit operations, and assess error propagation in the NOR using sensitivity studies such as Monte Carlo simulations (Figure 2).6 ,7
Process control strategy prerequisites. Any robust control strategy needs:
- Analysis of output process variables—key performance indicators (KPIs) and CQAs—of unit operations
- A controller that varies CPPs to achieve a robust and continuous Process
Some new at-line tools for CQA measurements use nuclear magnetic resonance spectroscopy or liquid chromatography–mass spectrometry.7 For continuous processing, however, they must be deployed as real-time PAT tools. Various solutions may bridge this task in the near future:
- Data-driven model-based approaches using spectroscopy
- Robust on-line sampling solutions that link gold standard analytics in online mode
- Model-based approaches that link CQAs to real-time measurements, offer a clear advantageous means to measure less
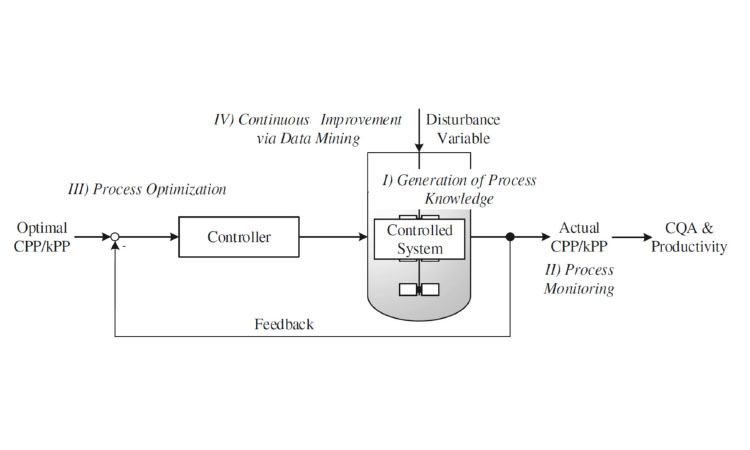
Developing a multivariate controller, which allows multiple CPPs as a function of multiple CQAs, is a more difficult challenge (Figure 3). Multiple-input/multiple-output controllers10 are available in other market segments, but are rarely applied in R&D bioprocessing labs. The trigger for this transition will be integrated real-time architecture that combines data management, real-time execution of models, advanced control algorithms, and model-generating workflows using “software as a service” tools.8 ,9
- 5
- 6 a b
- 7 a b Foley, D.A., et al. “Online NMR and HPLC as a Reaction Monitoring Platform for Pharmaceutical Process Development.” Analytical Chemistry 85, no. 19 (2013): 8928–8932.
- 10Kroll, P., et al. “Model-Based Methods in the Biopharmaceutical Process Lifecycle.” Pharmaceutical Research 34, no. 12 (December 2017): 2596–2613. https://link.springer.com/article/10.1007/s11095-017-2308-y
- 8Ulonska, S., et al. “Workflow for Target-Oriented Parametrization of an Enhanced Mechanistic Cell Culture Model.” Biotechnology Journal 13, no. 4 (November2017).
- 9Kroll, P., et al. “Workflow to Set Up Substantial Target-Oriented Mechanistic Process Models in Bioprocess Engineering.” Process Biochemistry 62 (November2017): 24–36.
Control Strategy Challenges
Fully connected, self-optimizing production processes, the first steps toward digital manufacturing, are well under way. Automation technology that leverages more meaningful data and analytics to improve processes (often referred to as manufacturing intelligence) will provide a better view of operations, better analytics, and real-time responsive decision-making to drive continuous improvement and operational excellence (Figure 4).
Process variability associated with critical material attributes (CMAs) arises from variations in raw material quality, variation in a supplier’s manufacturing process, and/or a change in suppliers. Combining ultraviolet high-pressure liquid chromatographic data with chemometrics can help determine variations in CMAs.
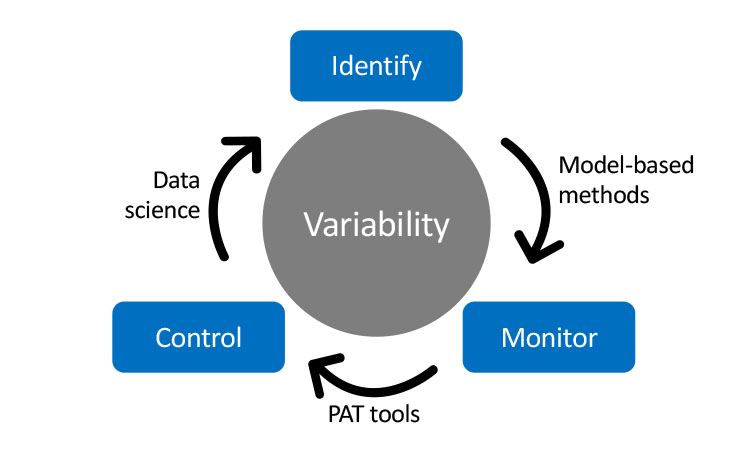
Key elements in drug development and product/process characterization are identifying and controlling CPPs and CMAs. These influence CQAs, which measure attributes associated with the safety and efficacy of the drug substance and drug products. We need to develop statistical methods to identify relevant CPPs and CMAs and approaches on how to control them to prevent adverse effects on drug product or drug substance (Figures 5 and 6):
- Get process understanding linking CMA to CQAs.
- Formulate this process understanding in a model, which allows control of the process while compensating for CMA variations.
- Future models will use “fingerprint” methods such as spectroscopy or chromatography as PAT to deploy the model in real time.
Variability in KPIs (such as cell growth) is of utmost importance for continuous bioprocessing development because it directly affects process performance and product quality. Identifying and controlling variability in KPIs is cumbersome and requires model-based methods.
Process modeling is employed to identify variations in process development. Automated workflows generate models in a structured frame, and then implement those models to identify sources of process variability. This requires real-time data collection, transmission, and analysis to capture process variability and predict process performance.
Equipment Design Challenges
In recent years, equipment manufacturers have introduced significant advancements in equipment design for CM applications in the biotech industry. The development of new technologies by the single-use industry have also increased the options for the biotech industry. While robust CM options still do not exist for all critical process steps in a typical biotech process, vendors continue to develop and improve their products.
Some technology allows true continuous processing with a steady-state approach; others use a staggered approach with units placed in parallel and fed alternately. These allow continuous flow in and out of the unit operation but do not reach a steady state. Table A summarizes the principle used for each unit operation. A typical process ow diagram for a fully continuous biotech process is shown in Figure 7.
Upstream Process
This section looks at the critical steps in a biotech process and evaluates the availability of CM equipment options currently on the market, focusing on equipment design. Cell culture is a very sophisticated process that requires stable parameters for advanced control algorithms to ensure an accurate response when a process perturbation occurs.
Current bioreactor system options are batch, fed-batch, and continuous (or perfusion). Batch, the classic option, is a closed system, and considered safer from a contamination perspective. It has the lowest product yield, however, since it requires four phases: lag (adjustment), log (logarithmic bacteria growth), stationary, and decline.
In a fed-batch system, continuous incoming substrates feed the reactor. By adding this fresh material, the volume of culture is greater, increasing product yield. Constantly feeding the bioreactor raises the contamination risk, however. From a design standpoint, this risk must be carefully studied to design a robust and reliable system that minimizes contamination risks.
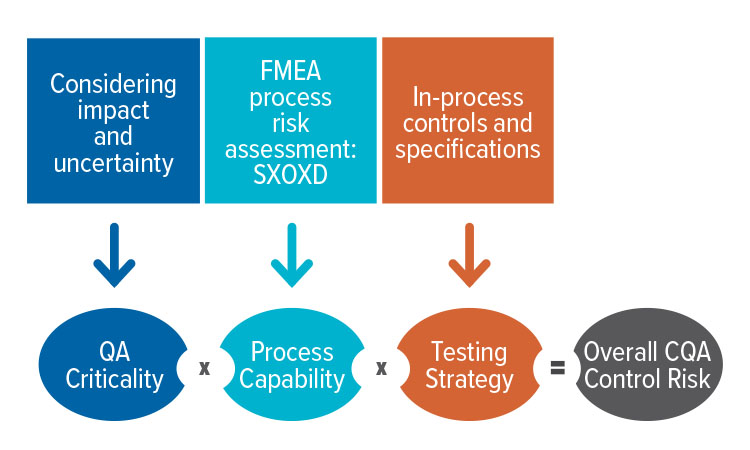
- Criticality of attributes and process parameters are needed to establish, understand, and evaluate a risk-based control strategy
- Testing strategy for any quality attribute (QA) depends on its criticality and process capability
The last option, a continuous or perfusion system, produces a higher product yield than either a batch or fed-batch system, while using the same size bioreactor. The process continually adds fresh media to the bioreactor while removing spent media and new product, keeping the bioreactor in either a lag or log growth phase. Bioreactors for continuous process applications are widely available from a variety of equipment manufacturers, with sizes ranging up to 2,000 liters; systems can also be customized for specific needs. It seems clear that the industry will move forward with this technology, but some challenges must be addressed to ensure the process remains safe.
The first challenge will be the capital investment of the installation. Batch systems have a more manual operation than perfusion systems, and are more attractive from an initial investment standpoint. An economic study should be considered to make sure the investment is worth it for the process and product (or products) that will be manufactured. The batch size and the number of batches per year will dictate whether the continuous reactor suits the process.
Implementing any continuous operation requires automated controls to react to and adjust critical parameters in real time. PAT requires good definitions of critical parameters to monitor and adjust in a timely manner according to the ongoing operation. It is especially important for continuous processes that these controls are tuned for fast response to ensure high product quality and yield.
Another challenge is the risk of contamination, which is higher for continuous processes than fed-batch systems. Bioreactors are considered open systems; each time the system is opened to re¬fill feed or harvest cells, the risk of contamination rises.11 The main issue is detecting possible contamination in real time to reduce product losses. Installing the right instrumentation will help to detect potential contamination, but this remains a challenge for a continuous operation.
Downstream Process
Defiltration/ultrafiltration (DF/UF) systems are probably the most common process steps in the biotechnology industry for upstream and downstream processing. This process has been widely used for many years in batch operations, but as perfusion bioreactors are increasingly used in the industry, the process bottleneck has moved downstream. Combining continuous UF/DF with tangential flow filtration (TFF) prevents buildup and clogging that might otherwise occur.12
Currently, only a few suppliers offer continuous TFF, even though the technology is present throughout the pharma and biotech industries. Indeed, a reverse osmosis system that generates purified water in a continuous operation is proof that the industry is not far away from getting more alternatives for their continuous UF/DF systems. As off-the-shelf options for this technology remain scarce, the main challenge for manufacturers who wish to implement continuous TFF is to rely on custom solutions to meet a specific throughput and adapt to a specific product.
- 11Cellab GmbH. “Cell Cultivation in Closed Systems.”Cellab News, 16 December 2014. https://www.cellab.eu/en/news/news-archive/8-2014-12-cell-cultivationin-closed-systems.html
- 12Challener, Cynthia. “Evolving UF/DF Capabilities.”BioPharm International 31, issue 1 (1 January 2018):24–27. http://www.biopharminternational.com/evolving-ufdf-capabilities
As CM takes hold in the industry, vendors have started to develop continuous UF/DF equipment options. These solutions are often single-use, scalable systems that can be used for buffer exchange for final products, desalting or buffer exchange before or after column chromatography, as well as small-molecule contaminant removal.
Unlike other critical process steps, however, there are a limited number of continuous equipment options from a limited number of vendors for UF/DF. Since batch UF/DF are economical solutions, it is important to carefully evaluate whether the benefits of the continuous equipment outweigh the investment in the equipment and in the process development.
Chromatography
The chromatographic step in a biotech process can be an obstacle when converting from a traditional batch to a continuous process, often due to the long contact time required between adsorbent and adsorbate. One new technology to reach the market is continuous multicolumn chromatography, which is based on a staggered processing approach. Studies show these systems require smaller column volumes and reduced buffer volumes compared to a standard batch process, while producing the same amount of product. Additionally, they greatly reduce the cost of chromatographic media without altering the sorbent or buffer used in the process. By switching from a single to a multicolumn chromatography system, users gain great flexibility and greater productivity. See “Transitioning to Multicolumn Chromatography” in Pharmaceutical Engineering, May-June 2018, for a more detailed look at the challenges and results of implementing multicolumn chromatography systems in the biotech industry.13
Inactivation
Currently, there is no truly continuous equipment available for the inactivation process step. The main challenge in designing equipment able to perform this step continuously is the long residence time. Commercially available systems are based on a staggered batch process in which several mixers are fed alternately, pH is adjusted, residence time met, and product unloaded. This approach allows product to be fed continuously, but outfeed is not consistent. The coiled flow inverter, a new technology currently at the proof-of-concept phase, uses flow design principles similar to those used in a heat exchanger. It operates at laminar flow velocities and uses radial mixing to ensure homogeneity.14
Buffer and Media Preparation
Currently, large batches of bugger and media are prepared of-line and fed to each unit when in operation. For a continuous process, in-line dilution would help keep the storage volumes low. Batches of concentrated media and buffer sufficient to supply the continuous processing time (or a portion of it if the processing time is long) would be prepared off-line and diluted to the appropriate concentration for each unit. This type of design would be considered semicontinuous, as the bags of concentrated buffers would have to be changed when they were empty. This technology is currently available and ready to implement.
As with the upstream process steps, converting from a batch to continuous process poses challenges. End users must carefully evaluate their process and perform an economic analysis. In general, CM equipment has a higher capital investment because of the automation cost. An analysis could show that reduced facility, scale-up, and operational costs outweigh the increase. While the technology exists in the industry, it has not been fully developed for the biotech unit operations.
Bioburden
Another challenge is bioburden control. While the current batch downstream process is not sterile, bioburden is controlled. With increased run time, however, bioburden can build up in the process train. It is necessary to control the microbial load so that it does not exceed the limits that can be removed by sterile filtration. Converting to a more sterile process would eliminate this issue, but would result in a significant cost increase.
- 13Arnold, Lindsay. “Transitioning to Multicolumn Chromatography.”Pharmaceutical Engineering 38, no. 3(May-June 2018): 36–39.
- 14Klutz, S., et al. “Continuous Viral Inactivation at Low pH Value in Antibody Manufacturing.” Chemical Engineering and Processing: Process Intensification 102 (April 2016): 88–101.
A filtered, concentrated buffer solution and closed processing are concepts that can be used in process design to mitigate this issue. Studies focusing on filter membrane saturation and chromatography efficiency will help determine appropriate continuous batch run time. A charging, cleaning, and sterilization strategy could also be developed to increase the run time. Buffer tanks in between unit operations are an option, but do not align with continuous design philosophy. Equipment redundancy would allow the process to continue while another equipment unit is cleaned and changed over. This would also allow production to continue in the case of an equipment failure.
Validation Challenges
For all unit operations except perfusion, process robustness has not been formally demonstrated at a validated commercial scale. Once proof of concept has been established, development and engineering runs at a pilot scale will be necessary to validate the new technology. Studies focusing on the microbial load through time will be crucial to validate this process and make it an acceptable option.
When the technology has been developed and is ready to be implemented, a suggested methodology is:
Stage 1
Stage 1 validation and risk assessments are executed to design the control strategy as well as create user and functional requirements specifications (URS and FRS). The following information must be clearly defined in these documents:
- System boundary, including handshake points with upstream and downstream equipment, control system, and process utilities
- CQA and CPP target values measured and controlled by the equipment
- Product information and CQAs (e.g., sterility) not directly measured and controlled by the equipment
- Equipment-related risk mitigation measures defined in the risk-assessment phase
From these documents, test protocols to be executed during the commissioning and qualification (C&Q) phase (factory acceptance test; site acceptance test; installation, operational, and performance qualifications). Process-performance qualification can also be developed. A system-impact assessment that encompasses the facility and process utilities, control systems, and process equipment defines the criticality of each system in the manufacturing space. The level of testing required depends on whether the system has a direct or indirect impact on product quality. Because a continuous process functions as an integrated process, it is crucial to develop a testing strategy that will take this into account.
The main challenge in executing a C&Q strategy for a continuous process lies in the phasing and sequencing in which each system is started up and tested. The execution must match the process architecture, starting with individual components, their integration with control systems, and ¬ finally to upstream and downstream equipment as well as utilities. The validation master plan must lay out this integrated strategy (Figure 8).
Stage 2
Stage 2 process qualification is integrated in the lifecycle concept. A risk-based process design phase provides solid proof of reproducible commercial manufacturing. A process-performance qualification
strategy must choose a run time that represents a commercial batch and show how process variability is controlled and stable product is produced. A sampling plan that tests product homogeneity over time should be included.
The qualification strategy should include data integrity, since a fully automated process measures and acts on cGMP-critical data throughout the manufacturing process. This information must be secure, archived, easily accessible, and auditable.
Stage 3
In theory, the stage 3 validation phase in a CM process should be relatively easy to implement, since it is necessary to follow quality by design (QbD) principles to get to this point. An automated process with PAT provides the real-time data required to monitor CQAs and CPPs continuously. Variability within the process and product quality is detected and acted upon immediately. Overall trending and analysis on data can be implemented on multiple batches and provide the basis for continuous improvement.
Regulatory and Quality Challenges
Because continuous processes are new in the biopharmaceutical industry, one of the biggest challenges related to their implementation in commercial manufacturing is the lack of operational experience and the perceived difficulty around quality management. Two main points are often part of the discussions:
- Definition of a “batch” when a process is continuous
- Managing process deviations and product out of specifications
The FDA defines a batch or lot produced via a continuous process by a unit of time instead of a quantity of product (emphasis added):
- (2) Batch means a specific quantity of a drug or other material that is intended to have uniform character and quality, within specified limits, and is produced according to a single manufacturing order during the same cycle of manufacture.
- (10) Lot means a batch, or a specific identified portion of a batch, having uniform character and quality within specified limits; or, in the case of a drug product produced by continuous process, it is a specific identified amount produced in a unit of time or quantity in a manner that assures its having uniform character and quality within specified limits.15
This unit of time is defined by a variety of factors, such as:
- Commercial requirements
- Efficiency of unit operations impacted by time or quantity of product (i.e., filtration)
- Bioburden
- Leachables/extractables in single-use processes
The current quality strategy for manufacturing processes is based on segmenting operations with off-line quality checks between steps, and release management with quarantine status. For continuous processes, real-time release is more applicable as the continuous monitoring of CQAs ensure that quality is maintained throughout the manufacturing process.
The methodology for managing out-of-specification results must be determined during the design process, with various failure modes and effects analyses implemented in the QbD and subsequent quality approach. The process must react to variation at each stage and reject product when CQAs are not within specifications. These critical functions are qualified during the validation process. Additional offline verifications can also be implemented to verify the performance of these functions. As with a batch process, deviations must be properly investigated, and appropriate corrective actions implemented to correct the root causes.
The large amount of real-time data accumulated during the manufacturing process will provide confidence in the quality decisions made, but the systems in place must allow proper access and analysis of the data.
Conclusion
Continuous manufacturing aligns with Pharma 4.0 objectives by increasing productivity and product quality. A well-defined process in the R&D stage, integrated PAT, and fully automated processes are key to its successful implementation.
Five main challenges must be overcome for its successful implementation in commercial biotech manufacturing:
- Develop control algorithms
- Implement PAT technology
- Design continuous equipment for all unit operations
- Develop a strategy for bioburden control
- Overcome lack of operational experience
The regulatory framework allows for continuous processes in commercial manufacturing and current validation methodology can be followed. With CM’s many advantages, increased investment in development from drug manufacturers will help solve these challenges and make cGMP continuous biotech manufacturing a new reality.
- 15US Code of Federal Regulations. Title 21, Chapter 1, Subchapter C, Part 210, Section 210.3. “Definitions.”https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=210.3


