Viral Vector Platforms: Intersection of Facility and Program

Realizing the promise of any novel viral vector therapeutic depends on the innovator’s ability to constantly meet evolving program requirements set in the product’s preclinical; clinical; chemistry, manufacturing, and controls (CMC); and market strategies. A key enabler to success is establishing a robust yet nimble viral vector manufacturing platform that delivers high-quality product on time and in full while managing costs.
Many paths are available in designing, delivering, and operating a viral vector manufacturing facility. This article explores a foundational design decision process that developers face: choosing an adherent versus suspension cell culture platform. Design considerations shift with each progressive stage in the product life cycle. Navigating the tradeoffs well amid high complexity and uncertainty is vital to success.
Viral Vectors in Gene Therapy
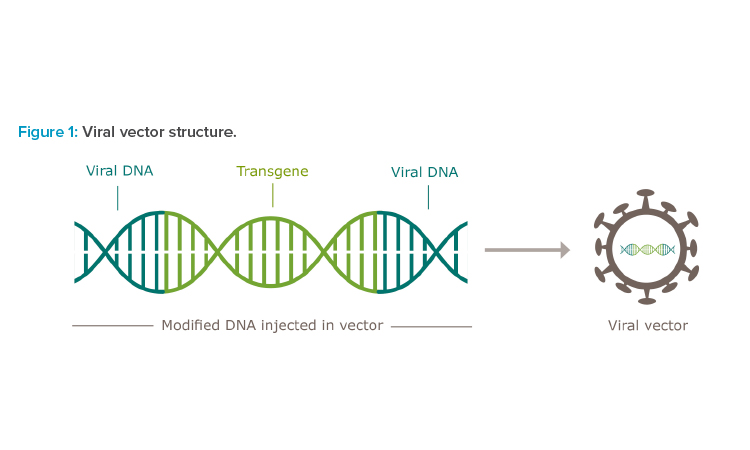
Human gene therapy seeks to modify or manipulate the expression of a gene or to alter the biological properties of living cells for therapeutic use.1 There are multiple approaches for gene therapy delivery, but the use of viral vectors is the most common.2 Viral vectors are viruses that have been modified to deliver a genetic payload to a cell while controlling the immunogenic and pathogenic risks associated with wild-type viruses (Figure 1).
Several types of viral vectors are commonly used in the pharmaceutical industry, each with their own tropisms, attributes, and features. Although an adenovirus vector has been predominant in the manufacture of COVID-19 vaccines, the most prevalent viral vectors used in gene therapy today are adeno-associated viruses (AAVs) and lentiviruses.
Though there are a number of notable differences between AAVs and lentiviruses—including genome type, size, and packaging capacity—the most notable distinction is that lentiviruses integrate into the host genome, whereas AAVs remain episomal. Integration into the host genome is associated with a higher risk of oncogenesis due to the risk of off-target integration.3 This in turn dictates the use of each viral vector type.
Because of their low immunogenicity, broad tropism, and nonintegrating properties, AAVs are typically used in therapies that are administered directly to the patient (in vivo therapies). Lentiviral vectors are predominant in therapies that are ex vivo, with the viral vector delivery occurring in cells that have been collected from the patient.4 With ex vivo therapies, lentiviral vectors can be safely used because the cells can be analyzed to confirm integration at the correct site prior to reintroduction into the patient.
![Figure 2: Gene therapy market for US clinical trial data through Q1 2022 [5].](/sites/default/files/2022-04/0522_PE_MJ_CoverStory_03.jpg)
Gene Therapy Market
The gene therapy pipeline is primarily focused on the oncological therapy area, with almost half the products in clinical trials focused on anti-cancer therapeutics, mainly lentivirus applications. Other product indications include rare diseases, neurology, and sensory therapy, which mainly utilize AAVs (Figure 2).
The gene therapy market continues to grow, with 2021 setting record numbers for funding. According to the American Society of Gene & Cell Therapy, the total number of gene therapies (including genetically modified cell therapies) in US clinical trials was 1,941 as of Q1 2022.5 The gene therapy market was expected to reach USD 3.42 billion by end of 2021 with a projected compound annual growth rate of 20.4% from 2021 to 2028.6
This sustained increase in demand for viral vectors presents a rising challenge across the pharmaceutical industry to develop laboratory and manufacturing facilities to supply the clinics and commercial markets with much-needed product.
Adherent vs. Suspension: Design Considerations
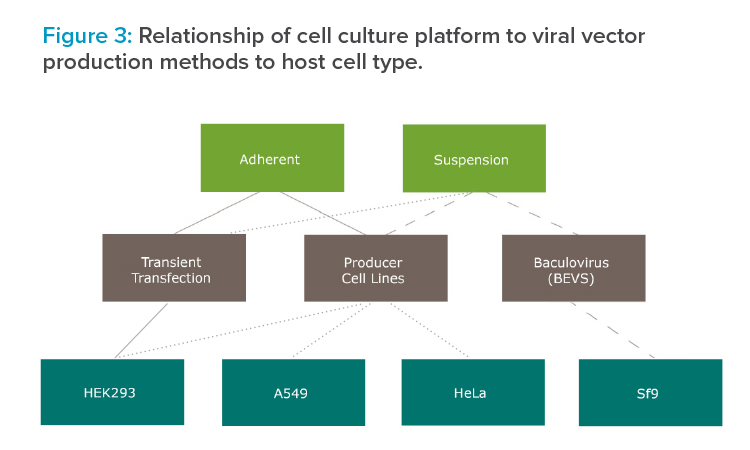
Figure 3 depicts the relationships between viral vector cell culture platform, viral vector production method, and host cell type.
Adapted Cell Lines
Gene therapy manufacturing employs a host cell line to produce viral vectors. Most host cell lines used for gene therapy manufacturing are human cell lines, although insect cell lines may also be used with specific technology platforms.
Human cell lines, which are developed from tissues, are inherently adherent in nature, requiring attachment to a solid surface in order to grow. In practice, this can be accomplished by using 2D platforms such as roller bottles or cell factories, and for 3D platforms with the use of microcarriers.
Suspension cell lines are cells that are grown floating in a culture medium with no anchorage dependence. Suspension cell lines are typically adherent cell lines that have been adapted to grow without dependence on a solid substrate. Elimination of serum-containing media is a step in the adaptation process from adherent to suspension. This step is beneficial because serum is a potential process contaminant.
Viral Vector Manufacturing Platforms
With both adherent and suspension cell lines, several different technology platforms are available for viral vector production. The most common meth-ods include transient transfection, use of producer cell lines, and infection of insect cells with baculovirus expression vector systems (BEVS). Other platforms are also available, but less prevalent in the pharmaceutical industry. The benefits and shortcomings of each platform are discussed next.
Transient Transfection
Currently, transient transfection is the most popular method7 of viral vector production because of its ability to introduce genetic material into the host cell line without the need to reengineer the host cell line for each distinct product.
Genetic material is introduced into the host cell line (typically a human cell line such as HEK293 cells) using multiple plasmids, each of which delivers critical material for the formation of a complete viral vector capsid. The genes are divided among multiple plasmids to prevent the formation of replication-competent viral particles through recombination. Typically, three-plasmid systems are used, although two- and four-plasmid systems are also options.
Higher volumes present challenges. Although transient transfection is effective at smaller volumes, when production is scaled up, transfection efficiency tends to decrease. This effect is a function of multiple factors: volume of transfection mixture (ratio with culture volume in bioreactor), mix time (overmixing can be detrimental), mixture incubation time prior to addition to the bioreactor, addition rate into the bioreactor, and various bioreactor conditions.
Producer Cell Lines
With the use of producer cell lines, the genetic material required to produce the viral vector is engineered into the host cell. Similar to transient transfection, a human cell line is typically used, although in this case it could be a HeLa or A549 cell line as well as HEK293 cells. Co-infection with a helper virus may be required to engineer a complete viral vector capsid.
Although producer cell lines are expected to deliver higher titers than transient transfection, there are downsides. A stable cell line that is engineered for the appropriate transgene must be engineered for each therapeutic treatment. Engineering and characterizing these stable cell lines require additional time and expense.
Baculovirus Expression Vector System
A third platform for viral vector production is the use of BEVS in Spodoptera frugiperda insect cells. Studies show that BEVS can result in up to a 10× increase in yield compared with production using transient transfection.8 This is largely attributed to BEVS being far less susceptible to fluctuations in environmental conditions in the bioreactor and the resultant inefficiencies.
With use of BEVS, baculovirus vectors must be engineered to deliver the therapeutic transgene as well as the viral vector structural components. A concern with using BEVS is that post-translational modifications may differ from human cell lines due to the use of insect cell lines.
Product Life Cycle in Program Development
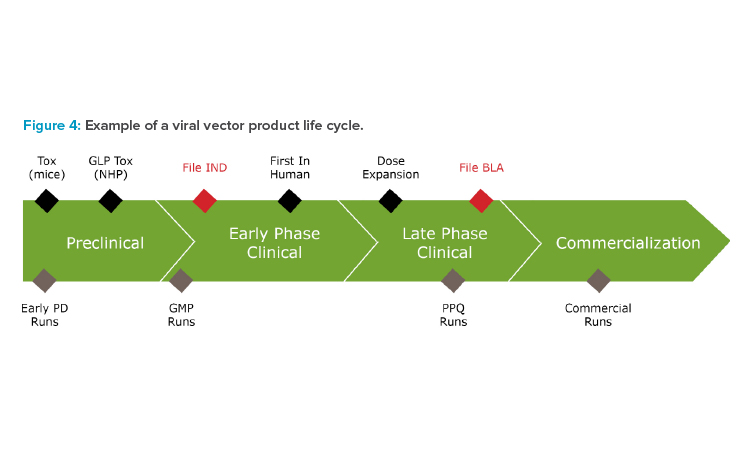
Innovators must heed the dynamic requirements of the product over its life cycle (Figure 4) to successfully execute viral vector programs. Decisions at every stage will critically impact subsequent stages and the success of the program.
Per an approved business case, the product must meet the sponsor’s regulatory, preclinical, clinical, and commercial program milestones. An integrated project plan draws a critical path across workstreams of process development, analytical development, stability, manufacturing, supply chain, quality, regulatory, and commercialization.
Amid high complexity, uncertainty, and changing conditions, the project team must act with agility and speed in making constant tradeoff decisions. It is imperative that they keep the patient’s needs central in satisfying the quality, safety, and efficacy guidelines in the quality target product profile.9
From early in the program, operational design criteria such as clinical and commercial process scale, supply chain capacity, process control strategy, manufacturability, and life cycle product cost should be factored in.
Viewing the entire product life cycle, we have observed several factors to consider for each phase when choosing an adherent or suspension viral vector platform.
Preclinical Phase
In the preclinical phase, a viral vector candidate is selected from a screen of dozens if not hundreds of permutations. Process understanding is limited, and associated challenges include high variation, low productivity, poor product characterization, immature analytical methods, and high process and product impurities.
Very small volumes of vector are commonly made from adherent cell lines in roller bottles or multilayer cell culture trays to support preclinical studies. At this scale, high-efficiency transient transfection is relatively fast and easy to develop, and is fit for purpose to keep pace with ongoing screening work and candidate selection.
In preparation for toxicology studies (tox), process developers must plot the course to either scale out 2D adherent technology (e.g., cell factories, fixed-bed bioreactors) or scale up and change to a suspension process. In any case, there are multiple benefits of having a tox process generating data early in the life cycle that is representative of the future commercial process.
For an ultra-rare gene therapy indication with relatively small anticipated commercial demand, adherent scale-out may be the best approach. However, for programs calling for high annual vector output, the use of adherent cells has volume constraints.10 A fixed-bed bioreactor can be used, though these do not exceed 600 m2 per unit. Another option is 2D cell factories. These are highly manual unit operations that can quickly scale to a tipping point where operational complexity and the related labor costs make this approach unfeasible for a commercial manufacturing facility.
Here, a switch to suspension culture may be viewed favorably due to its scalability. As noted previously, this change adds the burden and risk of having to adapt the cell line to a serum-free medium. This is a complex and lengthy process, and success is not guaranteed. It is common to find that cells adapted to suspension have a slower growth rate and may be genetically unstable.11 Once cells are adapted to growth in suspension, conventional single-use bioreactors can be used, ranging in size from 50 L to 5000 L, which is beneficial to operations. Stainless steel options are available at even larger volumes. This tradeoff decision is a pivotal one in the viral vector program.
Early and Late Phase Clinical
Informed by tox batch data, small-scale studies, and the developing clinical and market strategies, the GMP manufacturing process for human clinical trials can be locked down. The manufacturing and supply chain strategy defines a make versus buy approach, driving technology transfer of the process to either a contract and development manufacturing organization or an in-house GMP production facility. Analytical methods follow a similar track.
In the US, GMP manufacturing data generated will be included in the investigational new drug (IND) application. If the IND is cleared by the US Food and Drug Administration, this GMP viral vector will be used to dose patients in the first-in-human clinical trials and subsequent clinical trials. The manufacturing process supporting the late phase clinical trials must demonstrate consistency in the process performance qualification (PPQ) series where batches are run at commercial scale.
Through the clinical phases, it is ideal to stay the course with the chosen viral vector platform. However, major changes in viral vector demand may be discovered in the clinical phase. There may be multiple causes, ranging from clinical (e.g., high-end patient dose selected from the pivotal studies readout) to market (higher than expected patient adoption; accelerated plans to commercialize to new regions) to process (low cell culture titers and/or low downstream yield means fewer patient doses/batch at current scale). In this case, commercial supply cannot meet up-shifted demand, impairing the economic calculus of the baseline manufacturing strategy.
It is not unthinkable when heading into commercialization with the version 1 process that a parallel-path postmarketing process change might be prudent. This may mean a very late change from an adherent to a suspension platform, or from 500 L suspension to 1,000 L or even 2,000 L suspension. This decision is not taken lightly due to the heavy regulatory burden related to comparability requirements, technical risk, quality risk, potential delay, and the massive capital expense.
The project team must act with agility and speed in making constant tradeoff decisions.
Commercial Phase
With a successful PPQ series and related characterization work completed, the program prepares for a biological license application (BLA), product launch, and commercialization. With BLA approval and demand clearly in view, the process design and operational plans can be finalized with a manufacturability mindset.
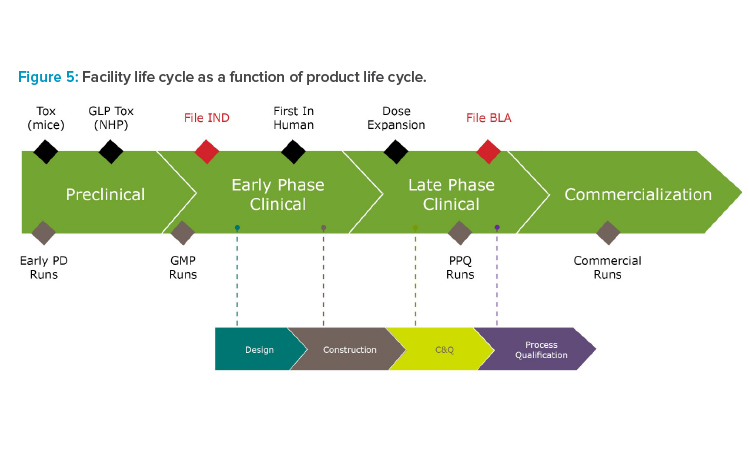
In considering the growth path to commercialization, sponsors may elect to construct a new GMP viral vector facility. Finding the right timing requires a delicate balance. Figure 5 depicts a scenario of an aggressive fast-to-market strategy, initiating a capital project facility build beginning in the early phase (phase 1/2).
While this can be a big win in removing lead time, it exposes the business to scope change risk. Finding the right investment trigger criteria means avoiding the risk of initiating too soon (with limited information) or too late (delaying time to market). Aligning capital project state gate decisions with product life cycle milestones (e.g., favorable clinical data readout) allows major capital investment projects to proceed with a more managed investment risk profile.
Investing in a feasibility study in the early design stage can be a beneficial mitigation tactic. A lot will change from the time of the original product business case. Stemming from the adherent or suspension platform approach, factors of scale and technology will help establish a specification for facility user requirements including an estimated required footprint, which is a good parametric predictor of overall facility capital cost.
Running a high-level comparative project financial evaluation of a commercial-scale adherent versus suspension solution sheds new light on estimated capital expense and operational/life cycle expense. Even if it is only an order of magnitude of precision, it can be a pragmatic time-saving approach.
Manufacturing Facility Considerations
In the manufacture of viral vectors, especially at larger scales, the application of suspension bioreactors may look similar to traditional biologics manufacturing. However, there are some unique facility design considerations for viral vector facilities.
Unlike monoclonal antibody facilities, which are typically classified as good large-scale practices or BSL-1, viral vector facilities are most often classified as BSL-2. With increasing biosafety levels, greater protective measures are required to protect both operators and the environment from potential exposure to infectious agents. This difference is driven by the use of human host cell lines (HEK-293, HeLa), helper virus (adenovirus), and the viral vector in production (lentivirus).
Additionally, containment is a critical aspect of viral vector manufacturing that drives the design of the facility layout, including unidirectional flow of personnel, materials, and waste as well as HVAC strategy. Viral particles are difficult to detect and eliminate if a breach occurs. Primary containment is achieved through the use of closed-unit operations, with secondary containment provided through air locking and HVAC design.
Unidirectional flow mitigates the risk of cross-contamination, especially in multiproduct facilities. However, it increases upfront capital expenditure costs, with features such as dedicated supply and return corridors, segregated incoming and exiting personnel, and material airlocks added to the overall facility footprint.
The HVAC strategy can include the dedication of air handlers to viral positive production areas, once-through air for critical processes, and pressurization schemes that provide for both containment and adherence to GMP manufacturing guidelines.
Downstream Design Considerations
Each approach in the production of viral vectors has a characteristic set of tradeoffs to consider for the design and operation when effectively pairing a GMP purification process train to the upstream process.
An important consideration that impacts facility design is upstream production bioreactor scale in relation to downstream technology. Because these factors are dependent on the maximum size bioreactor in use (either adherent or suspension) and the yield of that bioreactor, multiple upstream bio-reactor runs or batches may need to be pooled to have an efficient and GMP-capable downstream process.
Due to the longer duration and passaging time associated with cell culture as well as the current limited size of production bioreactors, it is not uncommon to have multiple upstream suites feeding a single downstream suite in viral vector manufacturing facilities. This may change in the future as technology improves and efficient vector production in larger single-use bioreactors becomes a reality.
For adherent cell lines in a fixed-bed bioreactor system, there is the benefit of retaining the cells within the single-use consumable. This leads to a reduction in the depth filtration required compared with a suspension-based platform, since less cell mass must be removed from the product.
A major purification challenge of note in viral vector manufacturing is the generation of empty capsids; that is, viral vector capsids that do not contain the gene of interest (GOI). Partial capsids can also form that contain truncated GOI vector or encapsidated genetic impurities. At very small scale, removing empty capsids and partially full capsids from the process stream is effectively performed via gradient density separation using ultracentrifuges. However, ultracentrifugation is a highly manual process that must be performed in small batches, and it is not a scalable technology. Moving forward, most manufacturers are adopting anion exchange chromatography as a scalable solution for the removal of empty or partially full capsids from the product.
Producer cell lines in AAV manufacturing require the use of a live helper virus such as adenovirus to co-infect the cells, thus necessitating an additional purification step to inactivate the live virus. Producer cell lines by design intend to improve vector production efficiency compared with transient transfection while removing the safety risk of residual plasmids as an impurity in the process stream.
Conclusion
Viral vector therapies are becoming increasingly popular for virtually every metric, from clinical trial applications to investment dollars spent on new facilities. As this industry grows and evolves, innovators must choose one of many paths to supply patients and markets with a safe and efficacious product to meet the rising demand.
Choosing the best manufacturing platform is far from straightforward. Central to this decision is understanding the advantages and disadvantages of applying either an adherent or suspension platform to your product and program. This compels product sponsors to create and execute a viral vector manufacturing strategy synchronizing technical, quality, and operational considerations based on the current product life cycle phase while anticipating future program and regulatory requirements. To make good decisions, one must constantly keep the end goal in mind: ensuring product safety, efficacy, and quality at every stage.
About the Authors




