The ideal case is a site using one cleaning procedure with an acceptable set of critical cleaning parameters (CCPs) for all products. However, some of the products manufactured in a topical drug facility can be difficult to clean. For example, the equipment for blending and packaging large-volume, high-viscosity formulations is complex, and the residues can be challenging to remove. Therefore, understanding and designing an effective cleaning process through laboratory testing, field trials, equipment designed for cleanability review, and risk assessments is critical to reducing resources and costs associated with cleaning validation and monitoring activities at the facility.
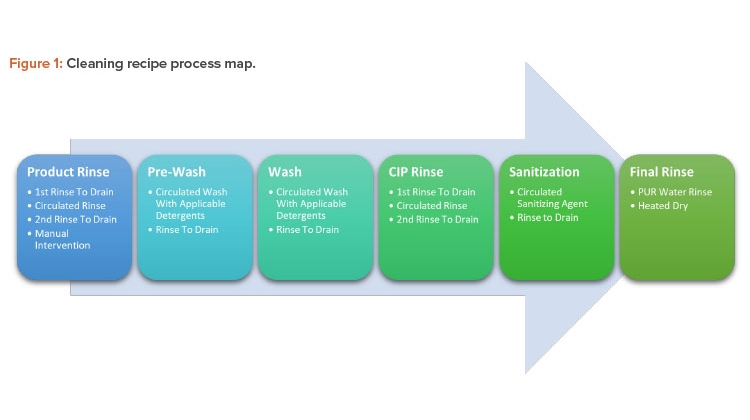
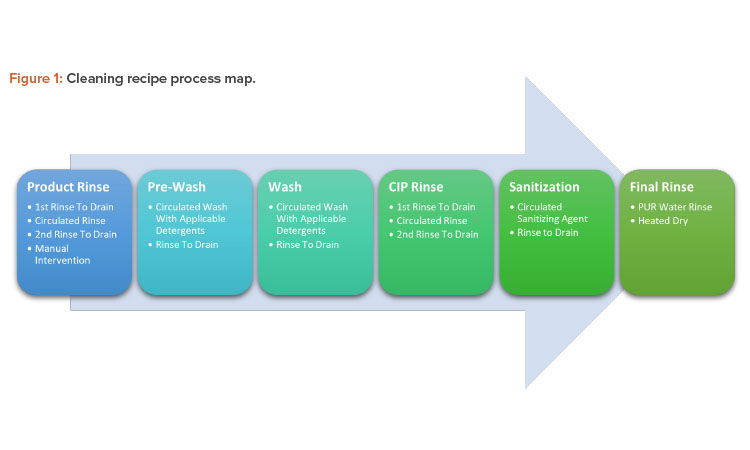
The cleaning validation life-cycle approach consists of three stages: design, qualification, and continued verification., Stage one: design, includes cleaning agents and suppliers, critical parameters and cleaning methods, laboratory and pilot testing, utility considerations, process equipment design review, cleaning process map, analytical test method validation, residue limits, visual inspection, and operations partnership.
Often referred to as the validation stage, stage two: qualification confirms that the cleaning procedure under normal conditions meets preestablished acceptance criteria. It includes cleaning validation master plan, product risk assessment, utility and equipment readiness, analytical method readiness, sampling site selection/grouping, standard operating procedures, validation protocols, execution of validation protocols, personnel training, and the validation documentation package. Stage three: continued verification includes periodic review, process control, continuous monitoring, preventive maintenance, and periodic revalidation (if applicable).
The cleaning process design involves reviewing the equipment, utilities, wastewater concerns, nature of residue, selection of cleaning agent, cleaning parameter, analytical method, and sampling method. Application of laboratory testing and field testing can be used to determine why selected conditions are used for the qualification stage but not the monitoring stage. Various CCPs may include, but are not limited to, cleaning concentration, temperature, wash time, water quality, surface material, and dirty hold time.
This article explores the life-cycle approach to cleaning topical drugs and cosmetics with attention to the cleaning design phase and leveraging this information, including lab studies and pilot runs, for qualifying and monitoring the cleaning process.
Cleaning Process Design
Cleaning Agents and Suppliers
Topical drugs and cosmetic products contain a wide range of components based on the desired properties of the products. These components can have poor solubility in solvents such as water and can be challenging to clean. Choosing an appropriate cleaning agent and supplier is critical to having an effective and efficient cleaning process. Like cosmetics, cleaning agents may be formulated chemistries that may contain several components to ensure broad effectiveness. They may, for example, contain components such as surfactants and chelants to help remove insoluble residues and drug actives to acceptable limits. It is also essential to understand the cleaning agent rinse profile, analytical methods for residue detection, and detergent toxicity profile. The supplier’s manufacturing process should provide a consistent product, quality expectations, and technical assistance to support the cleaning agent’s application within cGMP facilities.
Critical Parameters and Cleaning Methods
Laboratory testing is the first step in defining the critical parameters for removing process residues before materials are taken to the manufacturing floor. Critical parameters include time, action, concentration, and temperature (TACT), as well as water quality, surface, soil load and condition, and environmental factors. When designing the study, it is crucial to mimic plant conditions and restrictions as accurately as possible and to consider the method of cleaning. Most cleaning under consideration is for a clean-in-place (CIP) application, which consists of a spray impingement and cascading flow in the production vessel and turbulent flow through pipes and cleaning circuits.
The following procedure is used throughout the studies:
- Step one: Dry, clean 304 stainless steel coupons (7.5 × 15 cm size) with a 2B finish are weighed on an analytical balance (±0.1 mg) to obtain the pre-coating weight. This step establishes a baseline weight for the coupon, and will help determine how much residue should be coated on the coupon. In addition, the weight of the dried coupon will be used for comparison to all future weights after a simulated cleaning process.
- Step two: The coupons are coated with samples. The amount of residue per surface area is controlled and recorded.
- Step three: The conditioned coupon is weighed on an analytical balance to determine the pre-cleaning weight.
- Step four: Each coupon is cleaned with agitated immersion, spray wash, and cascading flow. Agitated immersion testing is usually the best method to determine cleaning chemistry, concentration, time, and temperature. The primary cleaning effect is caused by the chemical action of the cleaning agent. Spray wash is performed in a modified washer/disinfector or washer at 76 kPa (approximately 11 psi). Cascading flow is performed at 2 L/min (approximately 0.5 gal/min). These procedures simulate the expected cleaning process. If needed, higher pressure or flow rate is evaluated. If a spray ball apparatus is used for cleaning, spray impingement and cascading flow represent spray ball cleaning.
- Step five: Each coupon is removed at selected time points and visually observed for cleanliness.
- Step six: Each coupon side is rinsed with tap water for 10 seconds at a flow rate of 2 L/min. The temperature of the tap water is 20°C to 25°C unless specified otherwise.
- Step seven: Each side of the coupon is rinsed with deionized water and examined for a water-break-free surface. Water-break-free is a qualitative test that indicates the cleanliness of a metal surface. On a clean surface, free from organic residue, water sheets evenly—without any breaks in the water film as it runs from the surface of the metal panel.
- Step eight: Coupons are dried and then weighed on an analytical balance to determine the post-cleaning weight. The coupon is clean if it was (a) visually clean and (b) water-break-free, and (c) if its pre-coating and post-cleaning weights are equal (0.0 mg residue).
Laboratory and Pilot Testing
With multiple, complex products manufactured on the same equipment, the approach is to choose the most difficult to clean products to help narrow laboratory testing to approximately 25–30 samples. The primary benefit of lab testing is to provide a starting point for CCPs (action, temperature, time, and concentration). These data become the basis for the cleaning cycle development at the pilot scale. An additional benefit of lab testing is to enable the initial creation of a grouping strategy or product families for pilot-scale work. Because there are products with active pharmaceutical ingredients (APIs) in the families (FDA-regulated topical drug products), the grouping strategy helps in the later stages of this work’s validation phase.
A 125 L pilot unit of the Symex system is used for development work to enable scaleup to the production systems. Given the short development window of six months, a parent-child model is used for our product portfolio of more than 300 products. The 300 products have been narrowed down to about 60 selected products during the developmental phase and grouped into families. The goal in the design phase is to create a program that would allow for a seamless transition to validation and monitoring.
Utility Considerations
The system’s key utilities are city water (hot and ambient), purified water (ambient), steam (heating the system), chilled water (cooling the system), and compressed air (controlling valves). The cleaning cycle uses city water (process water) for the initial rinse, wash, and sanitization cycles. The post-sanitization cycle uses purified water to heat the system for the final vacuum drying step.
The system is piped for hot water and city water because some products respond better to a cold-water rinse. The city water and purified water are controlled to within 1% of the set point. The other key utilities—steam (40 bar) and chilled water—are controlled within a range of 5% around these values. Heating and cooling of the system are kept to within +/- 3°C of the set point. A clean compressed air pipe to the system controls the operation of multiple valves. The dosing of water is controlled within a 1% v/v cleaning solution range. Steam pressure is at 40 bar, and the chilled water temp is controlled within a 5% range of this.
Table 1: Laboratory cleaning trials.
| Sample |
Cleaner |
Cleaning
Method |
Concentration |
Time per
Cleaning
Method (min) |
Visual
Observations |
Water-
Break-
Free |
Temp (°C) |
Mineral
Sunscreen |
Formulated cleaner containing potassium
hydroxide and detergent additive |
AI, SW, CF |
3% v/v + 3% v/v |
45 |
Visually clean |
Yes |
80 |
| PGL Polymer |
Formulated cleaner containing potassium
hydroxide and detergent additive |
AI, SW, CF |
5% v/v + 5% v/v |
60/30/60 |
Visually clean |
Yes |
80 |
| Allianz OPT |
Formulated cleaner containing potassium
hydroxide and detergent additive |
AI, SW, CF |
3% v/v + 3% v/v |
60/30/45 |
Visually clean |
Yes |
80 |
Deodorizing
Body Spritzer |
Formulated cleaner containing potassium
hydroxide |
AI, SW, CF |
2% v/v |
15/3/30 |
Visually clean |
Yes |
60 |
Perfume
Compound |
Formulated cleaner containing potassium
hydroxide |
AI, SW, CF |
1% v/v |
10 |
Visually clean |
Yes |
60 |
Waterproof
Mascara |
Formulated cleaner containing potassium
hydroxide and detergent additive |
AI, SW, CF |
2% v/v + 2 % v/v |
60/45/60 |
Visually clean |
Yes |
60 |
| Foundation |
Formulated cleaner containing potassium
hydroxide and detergent additive |
AI, SW, CF |
2% v/v + 2% v/v |
60/30/60 |
Visually clean |
Yes |
60 |
| Serum |
Formulated cleaner containing potassium
hydroxide and detergent additive |
AI, SW, CF |
2% v/v + 2% v/v |
15/15/15 |
Visually clean |
Yes |
60 |
| Concealer |
Formulated cleaner containing potassium
hydroxide |
SW (52psi) |
3% v/v |
45 |
Visually clean |
Yes |
80 |
| Lip Gloss |
Formulated cleaner containing potassium
hydroxide and detergent additive |
SW, CF |
2% v/v + 2% v/v |
75/60 |
Visually clean |
Yes |
80 |
| Deep Cream |
Formulated cleaner containing potassium
hydroxide |
SW (52 psi) |
3% v/v |
15 |
Visually clean |
Yes |
80 |
| Foundation |
Formulated cleaner containing potassium
hydroxide |
SW (52 psi) |
3% v/v |
30 |
Visually clean |
Yes |
80 |
| Gel Cleanser |
Formulated cleaner containing potassium
hydroxide |
AI, SW, CF |
1% v/v |
15/15/15 |
Visually clean |
Yes |
60 |
Clear Proof
Face Mask |
Formulated cleaner containing potassium
hydroxide |
AI, SW, CF |
5% v/v |
90/30/60 |
Visually clean |
Yes |
80 |
Process Equipment Design Review
The pilot system has numerous capabilities within the primary operations of mixing, heating, cooling, induction, pressure, vacuum, and CIP. The main vessel for primary mixing and an auxiliary vessel for additional product phases or premixes are made of 316 SS with a specified roughness (RA) finish. The transfer from the auxiliary vessel to the main vessel can be done using a pump or a pressure differential between the tanks. The system has fixed piping except for the raw material induction, which uses flex hoses. The system was designed with sanitary fittings and gasket materials that are compatible with the cleaning chemistry.
The mixing elements consist of the central agitator, outer scrapers, and homogenizer, all of which provide the system’s flow and shear characteristics. There is an additional short and long loop in this system to enable batch turnover. The short loop goes from the homogenizer to the vessel’s bottom and the long loop enters about two-thirds of the way up the vessel. The induction ports for both the main vessel and phase vessel are located on the front panels.
From a capability standpoint, the phase vessel can induct liquids, powders, and pellets. The main vessel can induct via the liquid port (into the homogenizer well), the powder port (into the homogenizer well), and the bottom port (into the vessel bottom dome). This system can work in both manual mode (semi-auto) or automatic mode (auto). A batch recipe system allows creating programs for both batch making and CIP. The human–machine interface (HMI) is located on the floor level and the platform for ease of access and operation.
The CIP chemicals are added manually after the water is dosed. The homogenizer works as the pump for the system, circulating fluid to the spray balls. The phase vessel is fitted with two cascade-style spray balls, and the main vessel is fitted with four cascade-style spray balls and side spray jets. For the main vessel, the system controls the pressure to the spray balls via a set point with a maximum value of 2,500 mbar. The vessels were tested for spray coverage during the site acceptance test (SAT) using a riboflavin test. The flow through the secondary loops in the system, like the induction and transfer lines, is also controlled by the homogenizer speed and can be customized to ensure turbulent flow. During the development run, a borescope was used to inspect the lines to determine visual cleanliness. The internal walls and mixer blades were inspected visually using a flashlight.