Evolving China’s Regulatory System in Alignment with ICH

With the Chinese government initiating drug regulatory reform in 2015 and China joining the International Council for Harmonisation (ICH) in 2017, a significant number of measures have been implemented by the government. The aim is to make fundamental changes to China’s drug regulatory administration system so it can facilitate pharmaceutical development and better meet patient needs in the country.
Introduction to the Regulatory System in China
This article introduces these important regulatory changes, adaption of ICH guidelines, and key considerations for drug development in China, particu-larly those in chemistry, manufacturing, and controls (CMC), quality, and engineering areas. In addition, major challenges and opportunities will be discussed for innovative drug product development covering small molecules, biologics, and cell and gene therapy (C>) in China.
National Medical Products Administration (NMPA)
The State Administration for Marketing Regulation, which is directly under the State Council of China, administers the NMPA. The NMPA is China’s national regulatory authority for drugs, medical devices, and cosmetics. It was originally founded in 1998 as the State Drug Administration, joined ICH as a regulatory member in 2017, and was elected and reelected as a member of the ICH management committee in 2018 and 2021. The key functions and responsibilities of drug supervision are handled by internal departments and affiliated institutions under the NMPA during new drug clinical development and marketing authorizations.
The Center for Drug Evaluation (CDE)
The CDE is responsible for the acceptance and technical review of applications for drug clinical trials and drug marketing authorization. Two regional CDE centers are established in the Yangtze River Delta region and the Guangdong-Hong Kong-Macao Greater Bay Area to reach out to the pharmaceutical industry in these regions.
The National Institutes for Food and Drug Control (NIFDC)
The NIFDC is responsible for the sample testing and the method and specification verification according to the CDE’s review needs of new drug applica-tions (NDAs). The sample testing by the NIFDC is rarely required by the CDE for clinical trial applications, except for some vaccines and certain types of special biological products, e.g., some C> products. The NIFDC may allocate the testing and verification to the Provincial Institutes for Food and Drug Control for NDAs of small molecules.
The Center for Food and Drug Inspection (CFDI)
The CDI undertakes the preapproval inspection of the research and development site(s) and manufacturing site(s) as well as the for-cause inspection required by the CDE during the review of NDAs. It also coordinates the co-inspection if the GMP compliance inspection to the domestic manufacturing site(s) is initiated by
the Provincial Medical Products Administrations (PMPAs) together with the preapproval inspection. The CFDI may authorize the preapproval inspection to the PMPAs.
The Chinese Pharmacopoeia Commission (CPC)
The CPC publishes the national drug standards, including the Chinese Pharmacopoeia and other national drug standards. The national drug standards are mandatory for the drug products approved in China. Compliance with Chinese Pharmacopoeia is critical during the review, testing, and inspection of NDAs.
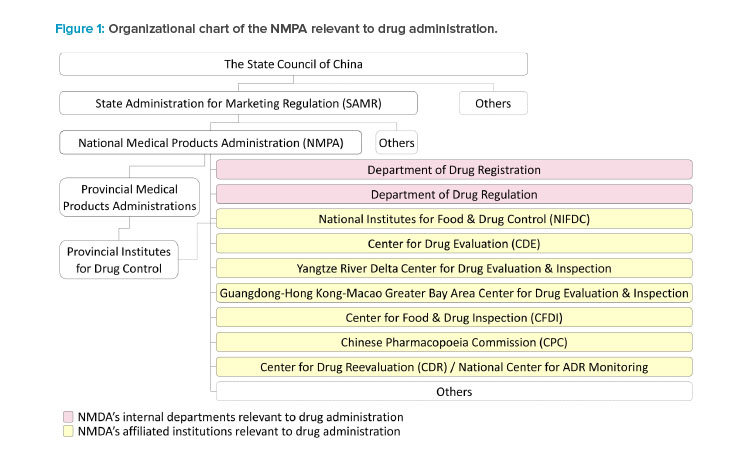
The Department of Drug Registration and the Department of Drug Regulation
These departments of the NMPA are responsible for interpretation of the laws and regulations to address the policy and regulatory and administrative issues raised by the CDE or the CFDI during the review and inspection of NDAs.
Provincial Medical Products Administrations (PMPAs)
Prior to the submission of a new drug application, the PMPAs are responsible for authorizing domestic manufacturing site(s) and the applicant that will be the marketing authorization holder (MAH) when the drug application is approved. PMPAs may initiate the GMP inspection together with the preapproval inspection of NDAs. Following approval of the new drug application, PMPAs supervise the drug production, distribution, pharmacovigilance, and most of the activities and obligations of the MAHs that take place at the PMPAs’ administrative areas.
Other Ministry-Level Departments
In addition to the NMPA, other ministry-level departments directly under the State Council are also relevant to drug research and development. For example, the ethics review of clinical trials is also subject to the relevant requirements supervised by the Ministry of Science and Technology (MOST) and the National Health Commission (NHC). The collection and research of human genetic resources during clinical trials need to be approved by the NHC (formally by the MOST), the human stem cell and gene diagnosis and therapy technology related drug clinical development is subject to the Special Administrative Measures on Access to Foreign Investment (Negative List) issued by the Ministry of Commerce (MOFCOM), and the technology exportation is subject to the Catalogue of Technologies Prohibited or Restricted from Export, issued by the MOFCOM as well.
Changes to Drug Administration Law and Implementation Rules
The Drug Administration Law (DAL)1 is the basis of drug administration in China, which was initially adopted in 1984 and revised in 2019 by the Standing Committee of the National People’s Congress. This revision consolidated the improvements and outcomes of the drug regulatory reform since 2015 and established the MAH-based regulatory supervision system throughout the life cycle of drugs. Specifically, the following improvements have been expected by the industry for a long time:
- Policies encouraging the development of innovative drugs with a new mechanism of therapy, for life-threatening or rare diseases, multitargeted system-atic intervention, and pediatric use.
- Conditional approval of drugs for life-threatening diseases without effective treatment or for urgent needs for public health, which are supported by predictable clinical value by available data on efficacy.
- Priority review and approval of drugs for pediatric use, drugs on shortage for urgent clinical needs, or new drugs for prevention and treatment of serious infectious diseases and rare diseases.
- Completion of clinical trial application review within 60 working days from the acceptance of application is stipulated, and expanded access and compassionate use of investigational drugs can be considered.
- MAH may have its drugs manufactured by their internal or contract manufacturing facilities, distributed by itself or by contactors; however, the MAH’s due obligations throughout the life cycle of drugs from research and development to use in patients are enforced and regulated.
| Regulatory Practices | Domestic Drugs | Imported Drugs* |
|---|---|---|
| Nationality of the applicant of clinical trial application and new drug application | Domestic sponsor | Foreign sponsor with its appointed local registration agent |
| Manufacturing authorization | The manufacturing license is granted to both the MAH and the manufacturer | N/A |
| Nationality of drug MAH | Domestic holder | Foreign holder with its appointed local agent |
| GMP compliance inspection | Inspected by PMPAs as part of routine supervision | Inspected by CFDI as the for-cause supervision called overseas inspection |
| Preapproval inspection of the manufacturing site(s) for new drug application | Initiated by the CDE and inspected by the CFDI or PMPAs for most new drug applications | For-cause initiated by the CDE and inspected by the CFDI for a few NDAs |
| Notification of moderate and above post-approval changes | Submitted to and reviewed by PMPAs | Submitted to and reviewed by the CDE |
| Review and approval of drug renewal | Submitted to, reviewed by, and approved by PMPAs | Submitted to, reviewed by, and approved by the CDE |
* For a chemical drug product, the imported drug means the drug product is produced at the foreign manufacturing site(s) and it must be held by a foreign entity (applicant/MAH). For a biological product, the imported drug means both the drug product and its drug substance(s) are produced at the foreign manufacturing site(s) and the drug product must be held by a foreign entity (applicant/sponsor/MAH).
Data Exclusivity and MAH in China
Based on the revision of the DAL, numerous regulations, standards, guidelines, and administrative working procedures have been updated or newly developed as a result of the drug regulatory reform. One of the most significant legislative programs is the draft revision of Regulations for the Implementation of DAL,2 with the following specific considerations for encouraging drug innovation and drug accessibility:
- Six-year regulatory data protection over the undisclosed trial data and other data of some drugs (to be further clarified by the authority) is expected.
- Up to seven-year market exclusivity for new drugs treating rare diseases is expected.
- Up to 12-month market exclusivity for pediatric-specific new drugs, new dosage forms, new strength, and the extension of pediatric indications or usage and dosage is expected.
- 12-month market exclusivity for the first generic drug succeeding in patent challenge is expected.
- Cross-border holding of marketing authorization is not yet allowed but may be expected in the future for innovative drugs with special considerations for urgent clinical demands.
The separation between the MAH and the manufacturer is one of the essential aspects of the MAH system regulated by the revision of DAL, in that MAH certificate of a drug product may be the same as the drug manufacturer or a different entity. However, due to the historical reasons, China’s drug regu-latory requirements and practices for imported drugs and domestic manufactured drugs are somewhat different (see Table 1). Both the MAH and the manufacturing site(s) must be from one side of the border; that is, an imported drug can only be held by an MAH outside of China, and a domestic drug can only be held by a domestic MAH.
Designations to Expedite Development
China has established three pathways in 2020 to expedite development and regulatory approval for medicinal products that have the potential to ad-dress unmet medical needs: breakthrough therapy, conditional approval, and priority review, which were described in the new Drug Registration Regulation.3 These pathways provide opportunities for developers to engage regulators during the development process and participate in accelerated review programs. Table 2 summarizes the qualifying criteria and key features for these three expedited pathways.
There is also another accelerated regulatory pathway called special review, which is available for public health emergencies, e.g., COVID-19 products.
Regulatory Interactions Between Health Authorities and Sponsor
The CDE has strengthened its resources to support innovation to ensure patient safety and efficacy while increasing patient access. Development of in-novative investigational products, such as C> products, can introduce unique challenges due to unknown safety profiles, complex manufacturing technologies, the incorporation of innovative devices, and the use of cutting-edge testing methodologies. In recognition of the complex nature of C> products, the CDE has introduced preliminary informal consultations to allow sponsors to obtain feedback to facilitate product development and clinical study planning. These early meetings are in addition to the conventional CDE and sponsor meetings. During the life cycle of drug development, sponsors may seek advice from the CDE regarding several topics, including, but not limited to, regulatory, clinical pharmacology, safety, product quality, and non-clinical subjects.
| Breakthrough Therapy | Conditional Approval | Priority Review | |
|---|---|---|---|
| Qualifying Criteria | • New therapeutics for condition with no current treatment options OR with major therapeutic advantage over existing treatments AND preliminary clinical evidence indicating the potential to produce significant benefits for patients with unmet medical needs and hence deemed “major interest” from a public health and therapeutic innovation perspective benefit | • New therapeutics fulfilling unmet medical need AND addressing seriously debilitating or life-threatening disease, rare disease, or for public health emergencies AND has a positive benefit-risk balance (benefits of immediate public access outweighs risks of incomplete data) AND it is likely the sponsor will be able to provide comprehensive data at anticipated time point in the future | • New therapeutics of major interest in terms of public health, particularly therapeutic innovation |
| Key Features | • Early dialogue to reinforce scientific or regulatory advice, optimize development, and enable priority review • CDE project manager appointed for a product at proof-of-concept stage • Kickoff meeting with multidisciplinary input from CDE experts to discuss development plan and regulatory strategy • Iterative scientific advice at major milestones • Advice on NDA preparation and submission • Use of priority review: procedure to speed CDE review | • Allows use of surrogate or intermediate end points to measure clinical benefit (i.e., tumor shrinkage versus overall survival) • Allows use of foreign clinical data when ethnically insensitive | • Reviews NDA in 130 days (versus 200 days for products without priority review) |
Generally, important milestone meetings include the pre-investigational new drug application (pre-IND), end-of-phase 1 (EOP1), EOP2, and pre-NDA) meetings. These meetings are categorized to three types of meetings related to the development and review of investigational new drugs and biologics: Type A, Type B, and Type C, the purpose and rule of which is very similar to the US Food and Drug Administration (FDA) meetings. For cutting-edge technology such as C>s, the CDE also provides pre-pre-IND meetings, similar to the US FDA INTERACT meeting, to address their consideration on nonclinical studies and CMC studies at the very early stage to help avoid uncertainty and waste of resources.
Pre-Pre-IND Meeting
Sponsors can obtain a preliminary informal non-binding consultation with the CDE through the pre-pre-IND meeting prior to a pre-IND meeting. Pre-pre-IND meetings are available for innovative investigational products at an early stage of development on issues that are not yet at the stage of pre-IND meeting. It is important to note that the pre-pre-IND meeting validates the CDE’s recognition of the complexity of such products.
Although a pre-pre-IND meeting is not mandatory, it may be highly valuable to drug development. This meeting is non-binding in nature, which means that a sponsor is not bound to pursue a particular regulatory pathway. This also means that the CDE feedback can change depending on infor-mation or updates the sponsor may provide in the future. Sponsors can obtain non-binding advice regarding different aspects during the development process, such as planning initial clinical development strategies, CMC, pharmacology/toxicology development, and clinical aspects of the product development program.
When seeking a pre-pre-IND meeting, identifying optimal timing for such a meeting relative to product development might be the sponsor’s greatest challenge. The meeting request might be declined if it is raised too early in the process at a point when a clear pre-clinical study design has not been established, or when it is considered too late, such as after a clinical development plan has already been defined. Nevertheless, sponsors are advised to apply for the pre-pre-IND meeting earlier rather than later because this meeting is the only opportunity to engage the CDE prior to the pre-IND process. If agreed, the CDE will hold the pre-pre-IND meeting within 60 working days upon receipt of the meeting request.
Pre-IND Meeting
The pre-IND meeting is a Type B meeting and meant to communicate the understanding of the action mechanism and discuss whether the available CMC, nonclinical data, or ongoing studies can support the early-phase clinical trials. The pre-IND meeting is not mandatory by regulation. However, in practice, it is expected if an IND involves the following topics: a product not previously approved, a new active pharmaceutical ingredient (API) with a novel pharmacologic mechanism, a product critical to public health, or a new indication.
| Regulatory Category | Category Explanations | Conditions |
|---|---|---|
| 1 | Innovative drugs not approved in and outside China | Drug substances and their drug products containing new compounds with definite structure and pharmacological actions and possessing clinical value. |
| 2 | Improved new drugs not approved in and outside China | 2.1 Drug substances and their drug products containing optical isomers with known active ingredients made through such esterification methods as separation or synthesis, or esterification of known active ingredients, or saltification of known active ingredients (including salts containing hydrogen bond or coordinate bond), or the alteration of the acid radicals, basic groups or metal elements, or the formation of other noncovalent bond derivatives (complex, chelate, or clathrate) and possessing significant clinical advantages. |
| 2.2 Drug products of new dosage forms containing known active ingredients (including new administration systems), new formulation processes, and new routes of administration and possessing significant clinical advantages. | ||
| 2.3 New combinations containing known active ingredients that can bring signifi cant clinical advantages. | ||
| 2.4 Drug products of new indications containing known active ingredients. | ||
| 3 | Drugs generic to original drugs approved abroad but not yet approved in China | Drug substances and their drug products possessing the same active ingredients, dosage forms, strength, indications, routes of administration and dosage, and method of administration as original drugs. |
| 4 | Drugs generic to original drugs approved in China | Drug substances and their drug products possessing the same active ingredients, dosage forms, strength, indications, routes of administration and dosage, and method of administration with original drugs. |
| 5 | Applications of drugs approval abroad for marketing in China | 5.1 Applications of original drugs marketed overseas (including drug substances and their drug products) for marketing in China. |
| 5.2 Applications of non-original drugs marketed overseas (including drug substances and their drug products) for marketing in China. |
Submission Content
Common Technical Document (CTD) or electronic CTD (eCTD)
The CTD or eCTD submission structure developed by the ICH provides the backbone for providing information regarding CMC, nonclinical, and clinical in structured modules. The CDE requires that all types of drug products use CTD or eCTD for IND and NDA.
The NMPA also stated that for clinical trial applications and marketing authorization applications for therapeutic biological products and preventive biological products, applicants should follow the “M4: Common Technical Document (CTD) for Registration Application of Drugs for Human Use”4 (hereinafter referred to as CTD) to prepare application dossiers.
Submission of IND and NDA to the CDE
Sponsors that wish to conduct a clinical trial or market product in China must submit an IND or NDA. The CDE’s review of the IND takes 60 working days. The CDE will focus on determining whether there are any reasons to believe the manufacturing or controls for the clinical trial product present unreasonable health risks to the subjects in the initial IND trials; as always, safety is the key concern for a reviewer. When filing an initial IND, details about the following CMC information are presented in the CTD structure and should include drug substance; drug product; placebo, if applicable; and labeling information for the labeled products relevant to the investigational drug.
If the CDE identifies any unresolved safety issue in the IND during the review, or if the CDE identifies such an issue arising during clinical study, the agency will issue a clinical hold on the clinical study. Regulations require the CDE to attempt to discuss and satisfactorily resolve any resolvable issue that may not result in the clinical hold with the sponsor before issuing the clinical hold. Once an IND has been deemed safe to proceed by the CDE, multiple studies can be conducted under the same IND, which are not limited to one specific indication at the early phase of development.
There are two main regulatory checkpoints: the clinical trial application (CTA) and the NDA. The CTA usually only requires the CDE’s review and approval decision. For the NDA, the CDE leads the other two functions (the NIFDC and CFDI) under the NMPA to make the technical decision and the NMPA makes the administration decision. During the review of the NDA of C> products, the NMPA’s CFDI has oversight of Good Laboratory Practice (GLP), Good Clinical Practice (GCP), and Good Manufacturing Practice (GMP) compliance and conducts on-site preapproval inspection. The NMPA’s NIFDC re-views the analytical method validation and tests the C> products per registered specification. All data and results from the CFDI and the NIFDC go to the CDE for comprehensive review and decision.
- 1“Drug Administration Law of the People’s Republic of China.” 26 August 2019. www.gov.cn/xinwen/2019-08/26/content_5424780.htm
- 2NMPA. “Regulations for the Implementation of the Drug Administration Law (Draft for Public Consultation).” 9 May 2022. https://www.nmpa.gov.cn/xxgk/zhqyj/zhqyjyp/20220509222233134.html
- 3“Drug Registration Regulation.” https://gkml.samr.gov.cn/nsjg/fgs/202003/t20200330_313670.html
- 4“Notice Regarding Implementation of the Level Two ICH Guidance (NMPA Order NO 51 2016).” 15 January 2018. https://www.nmpa.gov.cn/yaopin/ypggtg/ypqtgg/20180125175101686.html
Life Cycle Management
The NMPA established a three-level change management reporting system according to the risk level of changes. One of the challenges to innovation is the burden of global change management. Thus, the ICH developed the ICH Q12 Lifecycle Management Guidance: “Technical and Regulatory Considera-tions for Pharmaceutical Product Lifecycle Management,” which provides a framework to facilitate management of postapproval changes in CMC. The NMPA started implementation of the ICH Q12 guidance since 25 Aug 2023;5 and 24-month transition period has been set.
Chemical Drug Development
New Category of Chemical Drug
In March 2016, the NMPA announced the new chemical drug regulatory categories,6 as shown in Table 3. There are three key changes in the new category:
- The “innovative” definition changed from “not launched in China” to “not approved inside and outside of China.”
- The concept of Category II has similarity with 505b(2)7 in the US, but with emphasis that it should demonstrate “significant clinical value.”
- The “generic” requirements changed from “quality similarity” to “quality and efficacy should be similar to the originator.”
In June 2020, the NMPA8 further specified the regulatory category of chemical drug and the corresponding China submission document requirements should follow “ICH M4: The Common Technical Document,” signifying that ICH M4 was adopted in China.
Under the new regulation, because the “generic” concept has changed from “quality similarity” to “quality and efficacy similarity,” the NMPA initiated the “quality consistency reevaluation” program in February 2016,9 followed by the generic injection product “quality consistency reevaluation” program in May 2020.10 Drugs that pass the reevaluation program will have priority to be enlisted in the China National Essential Medicine List. Drugs that fail the reevaluation program will be removed accordingly.11 In addition, Chinese regulations required all the market products to have license renewal every five years, with generic product license renewals being rejected if it fails the reevaluation program.
Innovative Chemical Drug Development in China
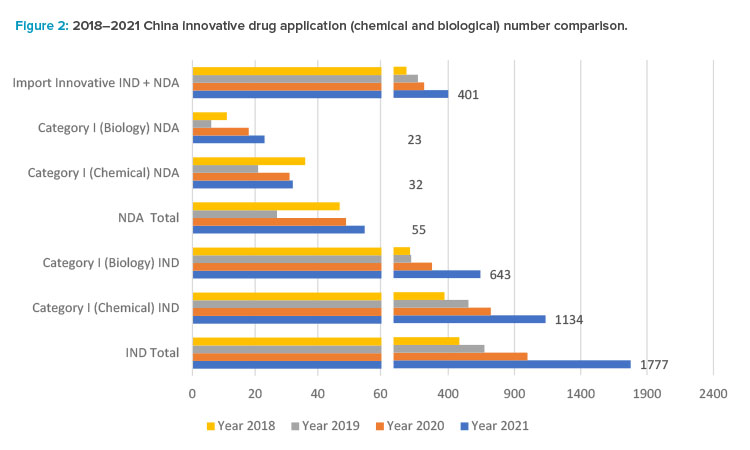
Small molecule drug product development has had a high growth rate in the past five years. Based on the 2021 CDE annual review report,12 1,886 innovative drug applications (IND and NDA) were accepted, with a year-on-year increase of 76%. Note that foreign companies contributed a significant number of these innovative drug applications.
Among all drug types, chemical drugs still dominate, and application numbers increased significantly: from 2,979 applications in 2017 to 6,788 applications in 2021, among which there are 1,166 innovative chemical drug applications (1,134 Category I INDs and 32 NDAs) belonging to 508 products, as one product may have different strength or dosage form, or different clinical trial applications. As revealed in the 2021 CDE annual report, there is a significant increase in regulatory applications with 35% in IND, 68% in NDAs, and 82% in Abbreviated New Drug Application (ANDA) compared to 2020.
China officially joined the ICH in June 2017. As of March 2023, 59 of 63 ICH guidelines in safety (S), efficacy (E), quality (Q), and multidiscipline (M) have been formally adopted. Among 151 ICH guidelines (including annexes and Q&As), 124 guidelines have been translated into Chinese and are available on the CDE website.
Major CMC issues are identified for generic products, including:
- There were serious defects in CMC, which cannot prove controllability for product quality.
- Proof for quality consistency between a generic and its reference drug was not established.
- Drugs used in different development stages, meaning that CMC variations were not comparable.
- Stability result and selection of starting materials for API did not meet technical requirements.
- Sourcing of APIs are not coming from validated supplier.
- Sample testing results were out of specification or analytical methods had serious deficiencies.
Adaption of ICH Guidelines
China officially joined the ICH in June 2017. As of March 2023,
59 of 63 ICH guidelines in safety (S), efficacy (E), quality (Q), and multidiscipline (M) have been formally adopted. Among 151 ICH guidelines (including annexes and Q&As), 124 guidelines have been translated into Chinese and are available on the CDE website.
For the quality series of guidelines, 17 ICH guidelines have been implemented in China, except for the following three guidance documents:
- Q4B Pharmacopoeias
- Q6B: Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products
- Q12 Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management
It is noted that Chinese Pharmacopoeia Committee has already put pharmacopoeia alignment with ICH as one of its top priorities.
Challenges and Solution
Chinese Pharmacopoeia
Different requirements by different pharmacopoeia are always an issue for global development. For example, because ICH Q4B13 and Q6B13 are not implemented in China, some test method differences (e.g., microbial enumeration tests, bacterial endotoxins test, and abnormal toxicity test) can pose issues for product registration in China. Moreover, differences in pharmacopoeia become critical under new registration regulation, which suggests that all sponsors need to complete registration sample testing before NDA submission; alternatively, the registration sample testing will be initiated by the CDE, which will make the registration process more unpredictable.
The sample testing issue may cause significant delays for the NDA approval. To deal with this issue, a CMC team should have a good understanding of Chinese Pharmacopoeia and relevant guidelines, which will assure that product specification meets the specific China registration requirements. This kind of challenge has been acknowledged by the health authority. The CPC plans to take a stepwise approach to adopting ICH guideline requirements in Chinese Pharmacopoeia 2025,14 including adopting ICH guidelines into general chapters of Chinese Pharmacopoeia; for example, adopting the ICH Q1 concept by revising the Chinese Pharmacopoeia stability general guidance; based on ICH Q3A13 and Q3B, 13 the Chinese Pharmacopoeia impurity analytical guidance has incorporated the reporting threshold, identification threshold, and qualification threshold concept, as well as the “Decision Tree for Identification and Qualification.”
Preapproval Inspection
Another challenge for innovative small molecule product development in China arose from Article 34 in the new Drug Registration Regulation,3 which required, “After having completed studies including CMC, pharmacological and toxicological studies and drug clinical trials supporting the drug marketing registration, established the specification and completed commercial-scale manufacturing process validation and manufacture for drug registration in-spection and testing, the applicant shall file a drug marketing authorization application and submit related study data as per application dossier re-quirements. The application shall be accepted when the application dossiers comply with requirements and pass the administrative check.”
It will bring challenges to the pharmaceutical company, because “completed commercial-scale manufacturing process validation” before NDA/marketing authorization application (MAA) submission means early CMC investment with more uncertainty and sacrifice of the shelf life of the vali-dation drug product batches.
It is not a major issue if the China NDA happens years later than another country or region, or if there is global simultaneous submission in multiple countries, including China. It will become a challenge if China is selected as the first country for the NDA. As a result, China-specific process validation requirements should be taken into consideration in a global CMC development plan.
| Regulatory category | Category explanations | Conditions |
|---|---|---|
| 1 | Innovative vaccines not approved in and outside China | 1.1 Vaccines for the disease without effective prevention methods. |
| 1.2 Neoantigen forms developed based on marketed vaccines, such as new recombinant vaccines, new nucleic acid vaccines, and new conjugate vaccines prepared based on marketed polysaccharide vaccines. | ||
| 1.3 Vaccines containing neoadjuvants or neoadjuvant systems. | ||
| 1.4 Multicombined/polyvalent vaccines containing neoantigens or neoantigen forms. | ||
| 2 | Modified vaccines developed by modifying domestically or overseas approved vaccine products | 2.1 Vaccines with obvious clinical advantages that are obtained by changing the antigen spectrum or type based on domestically or overseas marketed products. |
| 2.2 Vaccines with major technical improvements, including the improvements of the bacterial or viral strain/cell matrix/ production process/dosage form of the vaccine. | ||
| 2.3 New multicombined/polyvalent vaccines composed of marketed for similar vaccines. | ||
| 2.4 Vaccines with obvious clinical advantages that are obtained by changing the administration route. | ||
| 2.5 Vaccines with obvious clinical advantages obtained by changing the immunizing dose or immune procedure. | ||
| 2.6 Vaccines with changed applicable populations. | ||
| 3 | Domestically or overseas approved vaccines | 3.1 Overseas marketed manufacturing on oversea, domestically unmarketed vaccines to register for marketing. |
| 3.2 Overseas marketed and domestically unmarketed vaccines to register for domestic manufacturing and marketing. | ||
| 3.3 Domestically marketed vaccines. |
Biological Drug Development
Definition and New Category of Biologics in China
In 2020, the NMPA16 clarified the definition and classification of biological products as products that are manufactured from microorganisms, cells, animal- or human-derived tissues, body fluids, etc. as starting raw materials made by biological technologies, used for preventing, treating, and diagnos-ing human diseases. Biological products are further classified as three subgroups: preventive biological products, therapeutic biological products, and in vitro diagnostic reagents managed as biological products.
Preventive biological products are vaccines for human immunization to prevent and control the occurrence and prevalence of diseases, which are further divided as immunization program vaccines and non-immunization program vaccines (see Table 4). Therapeutic biological products are used for treating human diseases, including proteins, polypeptides, and their derivatives prepared from engineering cells (such as bacteria, yeast, and insect, plant, and mammalian cells) with different expression systems; C> products; allergen products; microecological products; biologically active products extracted from human or animal tissues or body fluids or prepared by fermentation (see Table 5).
In vitro diagnostic reagents that are biological products are regulated as therapeutic biological products, including in vitro diagnostic reagents used for blood source screening and radionuclide labeled in vitro diagnostic reagents.
It is noted that under the new regulatory classification system, a Class 1 new drug is defined as when the biologics license application (BLA)/NDA ap-plication is submitted in China and the product has not been marketed inside or outside China. Once the classification is determined, it will not be affected by product marketing authorization abroad. Therefore, this new registration classification system encourages simultaneous development and marketing authorization.
| Regulatory Category | Category Explanations | Conditions |
|---|---|---|
| 1 | Innovative biological products not approved in and outside China | Domestically and overseas unmarketed biological products for therapeutic. |
| 2 | Modified biological products developed by modifying domestically or overseas approved biological products | 2.1 Biological products with obvious clinical advantages that are obtained by optimizing the dosage form and administration route based on marketed products. |
| 2.2 Biological products with new indications that were not approved domestically and overseas and/or with changed applicable populations. | ||
| 2.3 New combination composed of marketed biological products. | ||
| 2.4 Biological products with major technical improvements made based on marketed products. | ||
| 3 | Domestically or overseas approved biological products | 3.1 Overseas marketed and manufactured in oversea, domestically unmarketed biological products to register for marketing. |
| 3.2 Overseas marketed and domestically unmarketed biological products to register for domestic manufacturing and marketing. | ||
| 3.3 Biosimilar. | ||
| 3.4 Other biological products. |
In 2022, the total acceptance number and approval number of NDAs for innovative drugs declined, but those corresponding numbers for biological products are on the rise, indicating the continued boom in the development of biological products in China.16
Introduction to the Development of Biological Products in China
The CDE annual report12 shows a boom in the regulatory applications for innovative biological products in China since 2017, reaching a peak in 2021 when CDE accepted 860 IND applications for biological products in 2021, up 48% from the previous year, including 643 IND applications for innovative biological products; and accepted 178 NDAs, up 41% from the previous year, including 23 NDAs for innovative biological products.
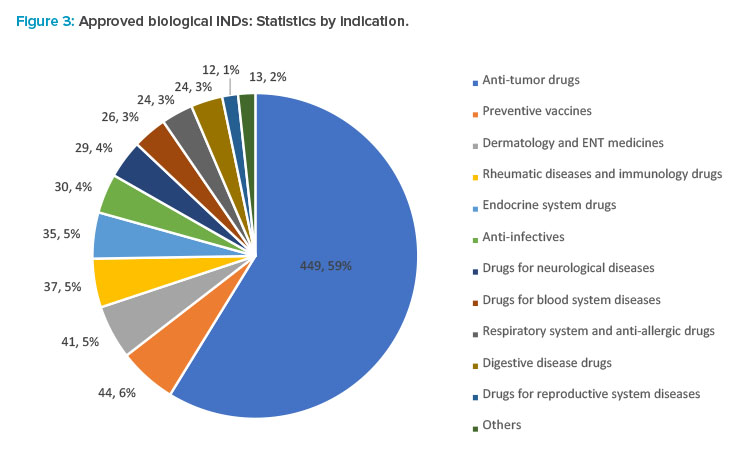
In 2021, the CDE recommended the approval of 764 IND applications for biological products, up 52.8% year from 2020, of which 449 were applications for anti-tumor indications. Applications for other indications included 44 for preventive vaccines, 41 for dermatology and ear, nose, and throat (ENT) medicines, 37 for anti-rheumatic and immunologic drugs, 35 for drugs acting on the endocrine system, 30 for anti-infective, and 29 for drugs for neurological diseases (see Figure 3).
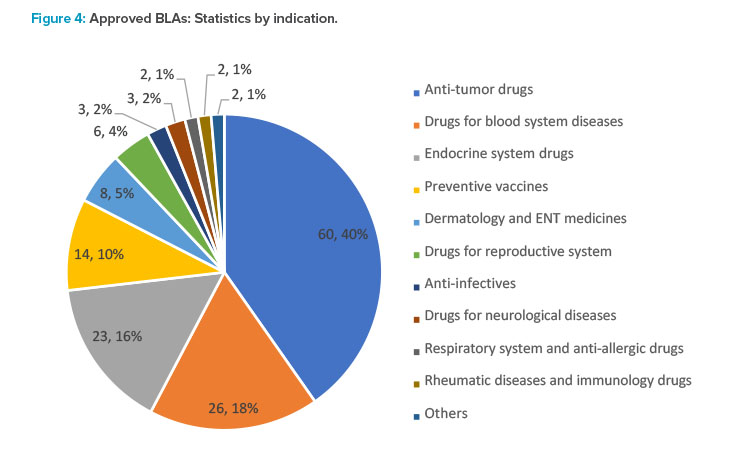
In 2021, the CDE recommended the approval of 149 NDAs, up 67.4% year from 2020, of which 60 were applications for anti- tumor drugs, making up the majority. Applications for other indications included 26 drugs for hematologic disorders, 23 for drugs acting on the endo-crine system, 14 for preventive vaccines, and eight for drugs for dermatology and five for sense organs (see Figure 4).
In 2022, the total acceptance number and approval number of NDAs for innovative drugs declined, but those corresponding numbers for biological products are on the rise, indicating the continued boom in the development of biological products in China.16
Implementation of ICH Guidelines and Harmonization of Other Related Regulations
Implementation of ICH Guidelines
As described previously, China has made significant progress in the implementation of and harmonization with ICH guidelines over the past few years, and the ICH Q5 series of guidelines13 related to biological products have all been implemented. For biological products with a global presence, the biggest challenge emanates from the harmonization of the Chinese Pharmacopoeia with ICH Q6B.13 How to scientifically explore and justify what test items should be included in the release specification based on the principles of ICH Q6B guidelines is currently a major issue for the development and approval of biological products in China.
- 5NMPA. “Notice Regarding Implementation of ICH Q12: Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management Guidance (NMPA Order NO 108, 2023).” 25 August 2023. https://www.nmpa.gov.cn/xxgk/ggtg/qtggtg/20230825171502197.html
- 6NMPA. “Notice of the Work Plan for the Reform of Chemical Drug Registration Classification. (NMPA Order NO 51 2016).” 9 March 2016. www.nmpa.gov.cn/xxgk/ggtg/qtggtg/20160309151801706.html
- 7“Federal Food, Drug, and Cosmetic Act (the Act), USA, Section 505(b)(2).” 21 USC 355 - New drugs (govregs.com)
- 8NMPA. “Notice of the National Medical Product Administration on Issuing the Requirements for Chemical Drug Registration Classification and Application Dossier (NMPA Order NO 44 2020).” 30 June 2020. www.nmpa.gov.cn/xxgk/ggtg/qtggtg/20200630180301525.html
- 9“Opinions of the General Office of the State Council on Conducting Quality and Efficacy Consistency Evaluation of Generic Drugs (NO 8 2016).” 6 February 2016. www.gov.cn/zhengce/content/2016-03/05/content_5049364.htm
- 10NMPA. ““Announcement of the State Food and Drug Administration on Conducting Quality and Efficacy Consistency Evaluation of Chemical Injection Genetics (NMPA Order NO 62 2020).” www.nmpa.gov.cn/zhuanti/ypqxgg/ggzhcfg/20200514162201667.html
- 11NMPA. “Notice of the National Medical Products Administration on Matters Related to the Evaluation of the Quality and Efficacy Consistency of Generic Drugs (NMPA Order NO 102 2018).” 28 December 2018. www.nmpa.gov.cn/xxgk/ggtg/qtggtg/20181228161301646.html
- 12 a b “2021 CDE Annual Review Report.” Released 1 June 2020. www.cde.org.cn/main/news/viewInfoCommon/f92b7bdf775bbf4c4dc3a762f343cdc8
- 13 a b c d e f ICH “Quality Guidelines.” https://www.ich.org/page/quality-guidelines
- 14“Outline for Compilation of the Chinese Pharmacopoeia (2025 Edition).” https://www.chp.org.cn/gjyjw/tz/17490.jhtml
- 3
- 16 a b c Essence Securities. “Innovative Drugs: Industry Structure Optimization, New Progress in Multiple Disease Fields and Internationalization is Worth Looking Forward To.” 6 February 2023. Biomedicine II.
In addition, how to resolve the contradiction with the Chinese Pharmacopoeia is also an issue posed to the implementation of ICH Q6B. For exam-ple, the tests for host cell protein residues and host cell DNA residues should be performed as required by “General Monograph of Recombinant DNA Protein Products for Human Use,” “General Monograph of Recombinant Monoclonal Antibody Products for Human Use” and other monographs concern-ing recombinant products in Chinese Pharmacopoeia.17 There are other issues for Chinese Pharmacopoeia to be harmonized with ICH guidelines, such as cell banks and viral safety with ICH guidelines Q5A, Q5B, and Q5D.13
Special Considerations for Vaccines
Quality Control of Vaccines
In general, testing for abnormal toxicity is required in the Chinese Pharmacopoeia, and more stringent limits are specified for residual process impuri-ties, such as inactivator, host cell protein and host cell DNA. Meanwhile, process validation and quality control are emphasized in the EU and US Pharmacopeias, and some testing items
(such as organic solvent residues and special immunities that can be effectively removed upon process validation) may be exempted; finished products may be exempt from some testing items (such as preservative content, residual solvent, protein impurities, and toxicity test) if intermediate products are qualified upon verification.
However, more emphasis is put on the final release testing for products in the Chinese Pharmacopoeia, and so there are more testing items for finished products than those defined in the European Pharmacopoeia (Ph. Eur.) and the US Pharmacopeia (USP). Finally, due to different identification methods, the requirement of the Chinese Pharmacopoeia for antigen content in vaccine products is higher than that of other pharmacopoeias.
Dossier Requirements for Vaccines
For vaccine clinical trial applications and marketing registration applications, dossiers should be prepared in accordance with ICH M4. The requirements for biological products in ICH M4 are mainly aimed for recombinant products. According to the characteristics of vaccines, there are more considerations regarding the CMC part of a dossier. 15
- If applicable, the data and information of virus used for production should be submitted in section 3.2.S.2.3.
- Verification reports of the batches of strains (virus) seeds used for production and the batches of cell matrix seeds used for production by the NIFDC or a third-party agency recognized by the drug regulatory agency should be provided in 3.2.S.2.3.
- For adjuvants, an overview of adjuvants should be submitted in 3.2.P; Comprehensive pharmaceutical research information should be submitted in 3.2.A.3, including raw materials, processes, quality attributes, testing methods, stability, etc.
- For the safety evaluation of exogenous factors, the target virus inactivation validation data should be submitted in 3.2.S.2.5. The validation data of re-moval/inactivation of non-target viruses should be submitted in the section 3.2.A.2.
Challenges
There are many challenges in the review and approval of biological products, with the biggest one perhaps from sample testing. According to the new Drug Registration Regulation, 3 registration sample testing is not required in an application for clinical trials of biological product, but subject enroll-ment for clinical trials of a vaccine product should be initiated only after vaccine product meets registration sample testing. In terms of NDAs, all biologics products are required to have three batches manufactured at commercial scale for registration sample testing, and the marketing approval may be granted after they pass the testing. It is undoubtedly a very challenging requirement for high-cost biological products, especially products for rare diseases.
Furthermore, attention should be paid to registration testing involved in supplemental applications for post-approval changes. As specified in the new Drug Registration Regulation, 3 the decision to initiate registration testing for major post-approval changes should be made based on risk assessment. However, the published regulations and regulatory documents are not clear about what post-approval changes entail the initiation of registration testing or what risk assessment model a decision-making is based on.
Moreover, once registration testing is initiated, the review timeline for major post-approval changes will be extended from 60 to 200 working days.18 Besides, the specific testing requirements are not clear, for example, sample size, batches, considerations for sample representativeness, and scope of the registration testing. It requires the applicant to discuss and reach an agreement with regulatory authorities on these issues in a timely manner, making post-marketing changes more difficult and uncertain.
In addition to sample testing associated with registration applications, imported biological products that have been granted marketing authorization are subject to import testing in compliance with the Administrative Measures for Import of Drugs19 issued in 2003. The customs may release such import biological products upon passing the testing prescribed in the registration specification. This requirement also increases the cost of drugs and prolongs the lead time for supply.
Another challenge is related to the segmented manufacturing and MAH cross-border holding. MAH is a new concept introduced in China’s regulatory framework in 2019. China health authorities are working on how to implement this concept to encourage innovation. There are many discussions about how the MAH can be held accountable for product quality, and segmented manufacturing is considered a high-risk practice.
Regarding the cross-border holding, at a practical level, the MAH, drug substance manufacturing site(s) and drug product manufacturing site(s) of im-ported biologics should all be located outside China. For example, except for insulins, it is not acceptable to ship drug substance to China and have drug product manufactured by a local factory, even if that factory is a member of the MAH group. Obviously, this is a big limitation for multinational corporations with global manufacturing networks and supply chains. However, given that the MAH system is still in its infancy in China, it is understandable that regulatory authorities still need to explore the most suitable management approach.
| Guideline | CMC | Nonclinical | Clinical | GMP | |
|---|---|---|---|---|---|
| 1 | Guidance for Clinical Trial Design of Gene Therapy for Hemophilia |
|
| X |
|
| 2 | Guidance for Clinical Trials of Immune Cell Therapy Products (Trial Version) |
|
| X |
|
| 3 | Guidance for Clinical Risk Management Plan for Marketing Authorization Application of Chimeric Antigen Receptor T Cells (CAR-T) Therapy Products |
|
| X |
|
| 4 | Guidance for Long-Term Follow-Up Clinical Studies of Gene Therapy Products (Trial Version) |
|
| X |
|
| 5 | Guidance for Nonclinical Studies of Gene Modified Cell Therapy Products (Trial Version) |
| X |
|
|
| 6 | Guidance for Nonclinical Studies and Evaluation of Gene Therapy Products (Trial Version) |
| X |
|
|
| 7 | Guidance for Research and Evaluation of Cell Therapy Products (Trial Version) | X | X | X |
|
| 8 | Guidance for CMC Research and Evaluation of Immune Cell Therapy Products (Trial Version) | X |
|
|
|
| 9 | Guidance for CMC Research and Evaluation of In Vivo Gene Therapy Products (Trial Version) | X |
|
|
|
| 10 | Guidance for CMC Research and Evaluation of In Vitro Gene Modified Systems (Trial Version) | X |
|
|
|
| 11 | Guidance for Cell Therapy Product GMP |
|
|
| X |
| 12 | Guidelines for CMC Research and Evaluation of Human Stem Cell Products (Trial) | X |
|
|
|
| 13 | Guidelines for Clinical Trials of Tumor Active immunotherapy Products (Trial) |
|
| X |
|
| 14 | Guidelines for CMC Research and Evaluation of Oncolytic Virus Products (Trial) | X |
|
|
|
| 15 | Guidelines for Clinical Trials of human stem cells and their Derived cell Therapeutic Products (Trial) |
|
| X |
|
Regulatory Framework for C> Products in China
In China, C> products are classified and regulated under the umbrella of biological products. Unlike the FDA Office of Therapeutic Products (OTP) or the EMA Committee for Advanced Therapies, the CDE does not have an integrated and special office or committee to provide oversight of the quality, safety, and efficacy. Instead, this is assessed by pre-clinical, biological CMC, clinical, and biostatistics and clinical pharmacology departments within CDE.
The novel and diverse nature of C> products has resulted in evolving regulatory activities specified to support these products. In the past three years, guidance documents addressing CMC, nonclinical, clinical, and GMP for C> were drafted and published by China authority (see Table 6). The list of guidelines initially constructed regulatory and technical requirements and is still growing.
As for the supervision of a clinical trial of C> products, China has two main regulatory authorities that are independent agencies with distinct roles—the NMPA/CDE, which supervises the clinical trial for registration purpose, and the NHC, which supervises the investigator initiated trial (IIT) for exploratory research purpose of new medical technology. IIT is mainly reviewed and approved by hospital’s ethics board and requires less CMC and non-clinical data than a CTA at NMPA/CDE. IIT pathway provides an opportunity for the sponsor to look at the efficacy and safety potential of their innovative C> designs before they invest more resources on CMC and nonclinical studies.
Considering C> is so innovative that regulatory framework is not as mature as small molecular and large molecular drugs, sponsors can obtain a preliminary informal non-binding consultation with CDE through the pre-pre-IND meeting prior to a pre-IND meeting.
Summary of C> China Regulatory Status
The regulatory framework governing C> products can pose a large degree of complexity for developers. In addition, in comparison to more traditional biopharmaceutical products, the field is relatively immature. Therefore, both development efforts and regulatory guidelines are evolving. CDE provides a range of opportunities for developers to meet, discuss, and gain clarification on various aspects of the C> product development process, including topics relating to early phases of development, CMC, and clinical trials.
In addition, references specific to C> products are starting to be addressed within the ICH.20 Materials developed and released by the ICH will provide guidance on a more general level and can be referenced as development progresses. Furthermore, CDE offers a number of expedited regulatory pathways to support and facilitate the innovation and therapeutic value promised by C> products. As the C> field continues to mature, it is expected that the corresponding regulatory structures and systems will continue to gain knowledge and experience from accumulated data, which will, in turn, allow both developers and regulators to move forward to ensure that patients are given access to the safest and most efficacious products possible.
Conclusion
The regulatory reform launched in 2015 has brought profound changes to the pharmaceutical industry in China and facilitated the adoption of ICH guidelines. As a result, innovative drug development has flourished in recent years, as evidenced by a significant number of small molecule biologics and C> products that have entered clinical development or gained marketing authorization in China. Nevertheless, technical or regulatory challenges present in pharmacopoeia, process validation, sample testing, or MAH should be overcome to support sustained drug development and patient accessibility. How-ever, we are confident that through dialogue, the regulatory authorities and the industry can work together to address those issues as the China regula-tory system continues to evolve in alignment with ICH.
- 17“Chinese Pharmacopoeia 2020, Volume III.”
- 13
- 15NMPA. “Notice of the National Medical Products Administration on Issuing the Requirements for Registration Classification and Application Materials of Biological Products (NMPA Order NO 43 2020).” 30 June 2020. www.nmpa.gov.cn/directory/web/nmpa/yaopin/ypggtg/ypqtgg/20200630175301552.html
- 3 a b
- 18NMPA. “Starting Work Procedure for Drug Registration Verification and Inspection (Trial) (NMPA Order NO 54 2021).” www.cde.org.cn/main/news/viewInfoCommon/c1dd9f7df30d686a2adab91f7f34587e
- 19“Import Drug Administration Regulation.” https://gkml.samr.gov.cn/nsjg/bgt/202106/t20210630_331800.html
- 20International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. “The ICH S12 Guideline Reaches Step 4 of the ICH Process.” News. 17 March 2023. www.ich.org/news/ich-s12-guideline-reaches-step-4-ich-process


